RHAPSIDO 25 mg
Rezumatul caracteristicilor produsului (RCP)
- 1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI
- 2. COMPOZIȚIA CALITATIVĂ ȘI CANTITATIVĂ
- 3. FORMA FARMACEUTICĂ
-
4. DATE CLINICE
- 4.1 Indicații terapeutice
- 4.2 Doze și mod de administrare
- 4.3 Contraindicații
- 4.4 Atenționări și precauții speciale pentru utilizare
- 4.5 Interacțiuni cu alte medicamente și alte forme de interacțiune
- 4.6 Fertilitatea, sarcina și alăptarea
- 4.7 Efecte asupra capacității de a conduce vehicule și de a folosi utilaje
- 4.8 Reacții adverse
- 4.9 Supradozaj
- 5. PROPRIETĂȚI FARMACOLOGICE
- 6. PROPRIETĂȚI FARMACEUTICE
- 7. DEȚINĂTORUL AUTORIZAȚIEI DE PUNERE PE PIAȚĂ
- 8. NUMĂRUL(ELE) AUTORIZAȚIEI DE PUNERE PE PIAȚĂ
- 9. DATA PRIMEI AUTORIZĂRI SAU A REÎNNOIRII AUTORIZAȚIEI
- 10. DATA REVIZUIRII TEXTULUI
1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI
Rhapsido 25 mg comprimate filmate
2. COMPOZIȚIA CALITATIVĂ ȘI CANTITATIVĂ
Fiecare comprimat filmat conține remibrutinib 25 mg.
Pentru lista tuturor excipienților, vezi pct. 6.1.
3. FORMA FARMACEUTICĂ
Comprimat filmat (comprimat)
Comprimat filmat rotund și curbat, de culoare galben deschis, cu diametrul de 6,7 la 7,6 mm, cu „LV” imprimat pe o parte și logo-ul companiei imprimat pe cealaltă parte.
4. DATE CLINICE
4.1 Indicații terapeutice
Rhapsido este indicat pentru tratamentul urticariei cronice spontane (UCS), la pacienții adulți care prezintă răspuns inadecvat la tratamentul antihistaminic H1.
4.2 Doze și mod de administrare
Tratamentul trebuie inițiat de medici cu experiență în diagnosticul și tratamentul urticariei cronice spontane.
Doze
Doza recomandată de remibrutinib este de 25 mg administrată oral, de două ori pe zi, o dată dimineața și o dată seara.
Dacă un pacient omite administrarea uneia sau mai multor doze de remibrutinib, trebuie sfătuit să ia următoarea doză la ora obișnuită. Nu trebuie administrate doze suplimentare de remibrutinib pentru a compensa doza sau dozele omise.
Medicilor prescriptori li se recomandă să reevalueze periodic necesitatea continuării tratamentului. Trebuie luată în considerare întreruperea tratamentului la pacienții care nu au prezentat niciun răspuns după 24 de săptămâni de tratament pentru UCS.
Întreruperea administrării dozelor
Se recomandă întreruperea administrării remibrutinib timp de 3 până la 7 zile înainte de o intervenție chirurgicală și timp de 3 până la 7 zile după o intervenție chirurgicală în funcție de tipul de intervenție și de riscul apariției hemoragiei (vezi pct. 4.4, 4.5 și 4.8).
Grupe speciale de pacienți
Vârstnici
Nu este necesară o ajustare specifică a dozei la pacienții vârstnici (cu vârsta ≥65 ani) (vezi pct. 5.2). La pacienții cu vârsta peste 65 ani, sunt disponibile date limitate privind utilizarea remibritinib.
Insuficiență renală
Nu este necesară ajustarea dozei la pacienții cu insuficiență renală (vezi pct. 5.2).
Insuficiență hepatică
Nu este necesară ajustarea dozei la pacienții cu insuficiență hepatică ușoară sau moderată. Remibrutinib nu este recomandat pentru utilizare la pacienții cu insuficiență hepatică severă (vezi pct. 5.2).
Copii și adolescenți
Rhapsido nu trebuie utilizat la sugari și copii cu vârsta sub 6 ani din cauza efectului posibil necunoscut asupra maturizării sistemului imun umoral (de exemplu, producerea de imunoglobuline și celule B cu memorie).
Siguranța și eficacitatea remibrutinib la copii și adolescenți cu vârsta cuprinsă între 6 și 18 ani nu au fost stabilite. Nu sunt disponibile date.
Mod de administrare
Administrare orală.
Remibrutinib poate fi administrat cu sau fără alimente. Pacienților trebuie să li se recomande să înghită comprimatul întreg, cu apă. Comprimatele nu trebuie rupte, sfărâmate sau mestecate pentru a asigura administrarea corectă a întregii doze.
4.3 Contraindicații
Hipersensibilitate la substanța activă sau la oricare dintre excipienții enumerați la pct. 6.1.
4.4 Atenționări și precauții speciale pentru utilizare
Risc hemoragic
La pacienţii cărora li s-a administrat remibrutinib au manifestat evenimente hemoragice uşoare până la moderate. Cele mai frecvente evenimente raportate au fost asociate cu echimoze, cum sunt peteşii şi contuzii (vezi pct. 4.8).
Pacienţii cărora li se administrează medicamente antitrombotice cu remibrutinib pot prezenta un risc crescut de sângerare. Riscurile și beneficiile administrării concomitente a medicamentelor antitrombotice cu remibrutinib trebuie luate în considerare (vezi pct. 4.5).
Pacienţii trebuie instruiţi să solicite sfatul medicului dacă apar semne şi simptome care sugerează sângerări semnificative. Dacă se suspectează o sângerare semnificativă, tratamentul cu remibrutinib trebuie întrerupt. După remitere, tratamentul poate fi reluat dacă se preconizează că beneficiul va depăși riscul.
Se recomandă întreruperea tratamentului cu remibrutinib timp de 3 până la 7 zile înainte de o intervenție chirurgicală și timp de 3 până la 7 zile după o intervenție chirurgicală, în funcție de tipul intervenției chirurgicale și de riscul hemoragic (vezi pct. 4.2).
Vaccinări
Siguranța remibrutinib administrat în asociere cu vaccinuri vii sau vii atenuate nu a fost studiată. Prin urmare, vaccinarea cu vaccinuri vii sau vii atenuate nu este recomandată în timpul tratamentului cu remibrutinib (vezi pct. 4.5).
Siguranţa remibrutinib administrat în asociere cu vaccinuri inactivate a fost studiată, prin urmare, în timpul tratamentului cu remibrutinib pot fi administrate vaccinuri inactivate. Pentru a optimiza răspunsul imun la vaccinurile inactivate, trebuie luată în considerare întreruperea tratamentului cu remibrutinib (de la 1 săptămână înainte de vaccinarea planificată până la 2 săptămâni după vaccinare) (vezi pct. 4.5).
Interacțiuni
Remibrutinib este un substrat al enzimei citocromului P450 3A4 (CYP3A4), prin urmare, există un potenţial de interacţiune cu alte medicamente administrate concomitent care sunt metabolizate de CYP3A4 sau care modulează activitatea CYP3A4 (vezi pct. 4.5).
Utilizarea concomitentă cu inhibitori potenți ai CYP3A4 creşte expunerea la remibrutinib şi, în consecinţă, poate creşte riscul de reacţii adverse la remibrutinib. Utilizarea concomitentă cu inhibitori potenți ai CYP3A4 trebuie evitată (vezi pct. 4.5).
Utilizarea concomitentă cu inductori moderați sau potenți ai CYP3A4 scade expunerea la remibrutinib și, în consecință, poate scădea eficacitatea remibrutinib. Utilizarea concomitentă cu inductori moderați sau potenți ai CYP3A4 trebuie evitată (vezi pct. 4.5).
Se recomandă monitorizarea mai frecventă a pacienţilor pentru reacţii adverse potenţiale atunci când remibrutinib se utilizează împreună cu substraturile glicoproteinei-P (P-gp) și ale proteinei de rezistență la cancerul de sân (BCRP), cu indice terapeutic îngust (vezi pct. 4.5).
Excipient cu efect cunoscut
Acest medicament conţine sodiu mai puţin de 1 mmol (23 mg) per comprimat filmat, adică practic „nu conţine sodiu”.
4.5 Interacțiuni cu alte medicamente și alte forme de interacțiune
Remibrutinib este, în principal, metabolizat prin CYP3A4.
Substanțe active care pot crește concentrațiile plasmatice ale remibrutinib
Inhibitori CYP3A4
Trebuie evitată administrarea concomitentă a remibrutinib cu inhibitori potenți CYP3A4. Administrarea concomitentă a ritonavir, un inhibitor potent al CYP3A4/P-gp, a determinat o creștere de 4,3 ori a ASC și o creștere de 3,3 ori a Cmax a remibrutinib.
Substanțe active care pot scădea concentrațiile plasmatice ale remibrutinib
Inductori CYP3A4
Trebuie evitată administrarea concomitentă a remibrutinib cu inductorii potenți sau moderați ai CYP3A4. Administrarea concomitentă a carbamazepinei (inductor potent până la moderat al CYP3A4) a scăzut expunerea sanguină a remibrutinib cu 74% (Cmax), respectiv 78% (ASC).
Substanțe active ale căror concentrații plasmatice pot fi modificate de remibrutinib
Substraturi transportoare/inhibitori
Se recomandă monitorizarea pacienților mai frecvent pentru a se identifica reacții adverse posibile la utilizarea remibrutinib cu substraturile P-gp și BCRP cu indice terapeutic îngust, mai ales când modificările minime ale concentrației pot duce la apariția reacțiilor adverse. Administrarea concomitentă a digoxinei (un substrat P-gp cu indice terapeutic îngust) în asociere cu remibrutinib a determinat o scădere de 1,4 a ASC și o scădere de 2,1 a Cmax a digoxinei. Administrarea concomitentă a rosuvastatinei (un substrat BCRP, fără indice terapeutic îngust) în asociere cu remibrutinib a dus la o creștere de 1,7 ori a ASC și de 1,6 ori Cmax a rosuvastatinei.
Într-un studiu de interacţiune medicamentoasă, efectul administrării remibrutinib (100 mg de două ori pe zi) asupra farmacocineticii midazolam (un substrat sensibil al CYP3A4) a dus la o creştere cu 43% a ASC şi la o creştere cu 27% a Cmax a midazolamului. Efectul dozei clinice de remibrutinib (25 mg de două ori pe zi) nu a fost studiat și poate fi diferit. Remibrutinib nu trebuie utilizat concomitent cu substraturi ale CYP3A4 cu indice terapeutic îngust (de exemplu, ciclosporină, tacrolimus, digoxină, warfarină, carbamazepină).
Contraceptive orale
Nu se anticipează că administrarea concomitentă a remibrutinib va avea un impact advers asupra eficacității contraceptivelor orale care conțin etinilestradiol și levonorgestrel (substraturi CYP3A4), dat fiind că expunerea lor nu a scăzut în prezența remibrutinib 100 mg de două ori pe zi (1,28, respectiv 1,36 ori creștere a Cmax și 1,16, respectiv 1,39 ori creștere a ASC).
Efectul remibrutinib asupra răspunsului imunitar la vaccinuri
Nu sunt disponibile date privind efectele vaccinurilor vii sau vii atenuate la pacienții cărora se administrează remibrutinib și aceste vaccinuri nu trebuie administrate concomitent cu remibrutinib (vezi pct. 4.4).
Pe baza unui studiu privind răspunsul imunitar la vaccinare, la voluntari sănătoși, pot fi administrate vaccinuri inactivate în timpul tratamentului cu remibrutinib. Pentru a optimiza răspunsul imunitar la vaccinurile inactivate, trebuie avută în vedere întreruperea tratamentului cu remibrutinib (de la 1 săptămână înainte de vaccinarea planificată până la 2 săptămâni după vaccinare).
Studiu privind răspunsul imunitar la vaccinare
Într-un studiu controlat cu placebo la voluntari sănătoşi care au administrat remibrutinib 100 mg de două ori pe zi, răspunsul imun la vaccinurile inactivate nu a fost afectat semnificativ atunci când remibrutinib a fost întrerupt cu 1 săptămână înainte de vaccinare până la 2 săptămâni după vaccinare. Cu toate acestea, tratamentul concomitent cu remibrutinib a fost asociat cu o reducere cu 60% a respondenților la vaccinul cu polizaharide PPV23 independent de celulele T, o reducere cu 21% a răspunsului IgG la vaccinul cu hemocianină (KLH) (neoantigen dependent de celulele T), rate de răspuns comparabile (1 până la 14% reducere) pentru 3 din 4 antigeni din vaccinul antigripal (dependent de celulele T) şi o reducere de 27% pentru 1 din 4 antigeni gripali.
Efectul remibrutinib asupra agenților antitrombotici
Nu sunt disponibile date privind administrarea concomitentă a remibrutinib cu medicamente anticoagulante. Trebuie avute în vedere riscurile și beneficiile administrării concomitente a agenților antitrombocitari cu remibrutinib (vezi pct. 4.2, 4.4 și 4.8).
Copii și adolescenți
Au fost efectuate studii privind interacțiunile numai la adulți.
4.6 Fertilitatea, sarcina și alăptarea
Femeile aflate la vârsta fertilă
Femeile aflate la vârsta fertilă, active din punct de vedere sexual, trebuie să utilizeze măsuri contraceptive eficace (metode care au ca rezultat rate ale sarcinii de sub 1%) în timpul tratamentului cu remibrutinib și timp de cel puțin 1 săptămână de la ultima doză. Femeile aflate la vârsta fertilă trebuie informate că studiile la animale au arătat că remibrutinib este nociv pentru fătul în dezvoltare (vezi pct. 5.3).
Sarcina
Datele provenite din utilizarea remibrutinib la femeile gravide sunt limitate. Studiile la animale au evidenţiat efecte toxice asupra funcţiei de reproducere (vezi pct. 5.3). Rhapsido nu este recomandat în timpul sarcinii.
Alăptarea
Nu se cunoaşte dacă remibrutinib/metaboliţii acestuia se excretă în laptele uman. Nu se poate exclude un risc pentru nou-născuți/copii mici. Alăptarea trebuie întreruptă în timpul tratamentului cu remibrutinib și timp de 1 săptămână de la administrarea ultimei doze.
Fertilitatea
Nu există date privind efectul remibrutinib asupra fertilității la om. Nu au fost observate efecte adverse asupra fertilității la șobolan, masculi și femele (vezi pct. 5.3).
4.7 Efecte asupra capacității de a conduce vehicule și de a folosi utilaje
Rhapsido nu are nicio influență sau are influență neglijabilă asupra capacității de a conduce vehicule și de a folosi utilaje.
4.8 Reacții adverse
Rezumatul profilului de siguranță
Reacția adversă cel mai frecvent raportată constă în infecții ale căilor respiratorii superioare (14,7%) cum sunt rinofaringită (6,6%) și gripă (2,5%).
Lista tabelară a reacțiilor adverse
Reacțiile adverse sunt enumerate în conformitate cu baza de date MedDRA pe aparate, sisteme și organe. În cadrul fiecărei categorii de aparate, sisteme și organe, reacțiile adverse sunt enumerate în funcție de frecvență, cu cele mai frecvente reacții adverse menționate mai întâi. În plus, categoria de frecvență corespunzătoare fiecărei reacții adverse este definită ca: foarte frecvente (≥1/10); frecvente (≥1/100 și <1/10); mai puțin frecvente (≥1/1 000 și <1/100); rare (≥1/10 000 și <1/1 000); foarte rare (<1/10 000); cu frecvenţă necunoscută (care nu poate fi estimată din datele disponibile).
Tabelul 1 Reacții adverse*
| Categorie de aparate, sisteme și organe | Reacție adversă | Frecvență |
|---|---|---|
| Infecții și infestări | Infecții ale căilor respiratorii superioare1 | Foarte frecvente |
| Infecții cu virusul herpes2 | Frecvente | |
| Tulburări ale sistemului nervos | Cefalee | Frecvente |
| Tulburări vasculare | Echimoze (Contuzii superficiale) Peteșii Contuzie3 Echimoză Purpură | Frecvente Frecvente Frecvente Frecvente Mai puțin frecvente |
Hemoragie Hematurie Epistaxis Hemoragie conjunctivală Hemoragie gingivală | Frecvente Frecvente Mai puțin frecvente Mai puțin frecvente Mai puțin frecvente | |
| Tulburări gastro-intestinale | Greață | Frecvente |
| Durere abdominală | Frecvente | |
| Tulburări musculo-scheletice și ale țesutului conjunctiv | Dorsalgie | Frecvente |
| Tulburări generale şi la nivelul locului de administrare | Pirexie | Frecvente |
* Studii de fază 3, controlate cu placebo, cu durata de 24 săptămâni, privind UCS 1 Infecțiile tractului respirator superior includ termeni preferați: infecție a căilor respiratorii superioare, sinuzită acută, sinuzită cronică, gripă H1N1, gripă, laringită, rinofaringită, faringită, faringită streptococică, faringoamigdalită, rinită, sinuzită, amigdalită, amigdalită bacteriană, infecție bacteriană a căilor respiratorii superioare, infecție virală a căilor respiratorii superioare 2 Infecții cu virusul herpes includ termeni preferați: herpes simplex, herpes zoster, herpes oral 3 Contuzia include termenii agreați: contuzie, tendința crescută de apariție a echimozelor, hematom | ||
Profilul de siguranță a remibrutinib la pacienții tratați timp de până la 52 săptămâni în REMIX-1 și REMIX-2 a corespuns reacțiilor adverse raportate în Tabelul 1.
Descrierea reacțiilor adverse selectate
Evenimente hemoragice mucocutanate
În perioada de tratament de 24 săptămâni, controlat cu placebo, în regim dublu-orb, din setul de date cumulate (studiile REMIX-1 şi REMIX-2, de fază III), evenimentele hemoragice mucocutanate (enumerate în Tabelul 1 la „Tulburări vasculare”) au apărut la 7,8% dintre pacienţii trataţi cu remibrutinib. Cele mai frecvente evenimente raportate au fost legate de echimoze: peteşii (3,8%) şi contuzii (2,3%). În general, la pacienții tratați cu remibrutinib, 92,0% dintre aceste evenimente au fost ușoare și 8,0% au fost moderate ca severitate. Timpul median până la debut a fost de 25 zile, iar durata mediană a fost de 22 zile. Toate cazurile s-au rezolvat spontan, fără tratament suplimentar. Nu s-a observat nicio asociere între evenimentele hemoragice mucocutanate şi numărul scăzut de trombocite. Administrarea concomitentă a remibrutinib cu anticoagulante nu a fost permisă în studiile clinice, dar a fost permisă administrarea concomitentă cu medicamente antiplachetare (acid acetilsalicilic (≤100 mg/zi) sau clopidogrel (≤75 mg/zi)) (vezi pct. 4.4 și 4.5).
La pacienții tratați cu remibrutinib, 0,5% au prezentat evenimente hemoragice mucocutanate care au dus la întreruperea definitivă a administrării remibrutinib și 1,0% au dus la întreruperea administrării remibrutinib (vezi pct. 4.2, 4.4 și 4.5).
Raportarea reacțiilor adverse suspectate
Raportarea reacțiilor adverse suspectate după autorizarea medicamentului este importantă. Acest lucru permite monitorizarea continuă a raportului beneficiu/risc al medicamentului. Profesioniștii din domeniul sănătății sunt rugați să raporteze orice reacție adversă suspectată prin intermediul sistemului național de raportare, astfel cum este menționat în Anexa V.
4.9 Supradozaj
În studiile clinice de fază I, nu au existat dovezi de evenimente adverse care să limiteze doza, observate la administrarea de remibrutinib în doze de până la 600 mg pe zi. Semnele și simptomele supradozajului cu remibrutinib nu au fost stabilite și nu există un tratament specific pentru supradozajul cu remibrutinib.
În caz de supradozaj, pacientul trebuie tratat simptomatic și trebuie instituite măsuri de susținere, după cum este necesar.
5. PROPRIETĂȚI FARMACOLOGICE
5.1 Proprietăți farmacodinamice
Grupa farmacoterapeutică: Imunosupresoare, imunosupresoare selective, codul ATC: L04AA60
Mecanism de acțiune
Remibrutinib este un inhibitor selectiv al tirozin-kinazei Bruton (BTK), care formează o legătură covalentă cu un reziduu de cisteină în situsul activ al BTK, ducând la inactivarea durabilă a BTK. Efectul terapeutic al remibrutinib în UCS se realizează prin inhibarea degranulării mastocitare și bazofile, inclusiv eliberarea histaminei și a altor mediatori proinflamatori, mediați de activarea patologică a IgE sau IgG îndreptată împotriva FcεR1 sau IgE.
Efecte farmacodinamice
Electrofiziologie cardiacă
Efectele remibrutinib asupra prelungirii intervalului QTc au fost anticipate utilizând analiza concentraţiei QTc. Limita superioară a intervalului de încredere de 90% pentru modificarea medie anticipată a QTcF, a fost sub 10 msec la Cmax anticipată la expuneri supraterapeutice. Prin urmare, nu este de așteptat o prelungire semnificativă din punct de vedere clinic a intervalului QTcF în cazul administrării terapeutice a remibrutinib.
Eficacitate și siguranță clinică
Eficacitatea și siguranța remibrutinib au fost evaluate în două studii identice, multicentrice, randomizate, în regim dublu-orb, controlate cu placebo, de fază III (REMIX-1 și REMIX-2) la pacienți adulți cu UCS inadecvat controlată, în ciuda tratamentului cu antihistaminice H1 de a doua generație.
În studiile REMIX-1 și REMIX-2, pacienții au fost randomizați într-un raport de 2:1 pentru a li se administra fie remibrutinib 25 mg, fie placebo, de două ori pe zi, pe cale orală, timp de 24 săptămâni, în timpul perioadei de tratament dublu-orb și în continuare într-o perioadă de tratament deschis de 28 săptămâni, în care toți pacienții au primit remibrutinib 25 mg, de două ori pe zi.
REMIX-1 şi REMIX-2 au înrolat un total de 925 pacienţi adulţi, diagnosticaţi cu UCS, care nu au fost controlaţi corespunzător în ciuda tratamentului cu o doză standard de antihistaminice H1 de generaţia a doua, definită prin prezenţa pruritului şi urticariei pentru ≥6 săptămâni consecutive. Toți pacienții trebuiau să aibă un scor săptămânal de activitate a urticariei (UAS7) ≥16 (interval 0 până la 42) și un scor săptămânal de severitate a pruritului (HSS7) ≥6 (interval 0 până la 21) timp de 7 zile înainte de randomizare. Pe lângă toți pacienții cărora li s-a administrat o doză stabilă de antihistaminic H1 de a doua generație (terapie de fond), pacienților li s-a permis să utilizeze un alt antihistaminic H1 de a doua generație, în funcție de necesități (terapie de salvare), la doze de până la 4 ori doza standard. Pacienţii au fost excluşi din aceste studii dacă au prezentat dovezi de boală cardiovasculară semnificativă din punct de vedere clinic, un risc semnificativ de hemoragie, tulburări de coagulare, infecţii cronice sau recurente, boală hepatică cronică sau acută cu semne de hepatită C sau B în curs, antecedente de boală renală, antecedente de sângerare gastrointestinală sau antecedente de malignitate în ultimii 5 ani.
Datele demografice și caracteristicile inițiale au fost, în general, bine echilibrate în toate grupurile. În studiile REMIX-1 şi REMIX-2, vârsta mediană a fost de 45 ani (interval: 18-79 ani) şi 41 ani (interval: 18-81 ani), cu 9,6% şi 7,7% ≥65 ani, și respectiv 68,3% și 65,3% pacienţi de sex feminin. Pacienţii au avut UAS7 mediu de 30,28 şi 29,99, ISS7 mediu de 14,59 şi 14,15 şi HSS7 mediu de 15,69, respectiv 15,84. La momentul inițial, 63,4% și 59,1% dintre pacienți au avut boală severă (UAS7 ≥28), iar respectiv 35,1% și 38,7% au avut boală moderată (UAS7 >16, și <28). 51,7% şi 46,6% dintre pacienţi au prezentat anterior angioedem în REMIX-1, respectiv REMIX-2. 68,1% şi 69,2% dintre pacienţi nu au administrat anterior anti-IgE biologic în REMIX-1, respectiv REMIX-2. Cel mai frecvent medicament biologic anti-IgE utilizat anterior a fost omalizumab (19,5% şi 19,0% în REMIX-1, respectiv REMIX-2).
Durata medie raportată a UCS la înrolare în toate grupurile de tratament a fost de 6,6 şi 5,2 ani în REMIX-1, respectiv REMIX-2, 39,4% şi 29,5% dintre pacienţi având o durată a UCS >5 ani.
Criteriul final principal de evaluare pentru studiile pivot a fost:
- modificarea absolută față de valoarea inițială a UAS7 în săptămâna 12.
Criteriile finale secundare de evaluare pentru studiile pivot au fost:
- modificarea absolută faţă de momentul iniţial a ISS7 şi HSS7 în săptămâna 12
- procentul pacienţilor care au atins un nivel bine controlat al bolii (UAS7 ≤6) în săptămânile 2 şi 12
- procentul pacienților care au prezentat absența completă a pruritului și urticariei (UAS7 = 0) în săptămâna 12
- procentul pacienților care au obținut scorul Dermatology Life Quality Index (DLQI) = 0-1 (da/nu) în săptămâna 12
- numărul de săptămâni cu control susținut al activității bolii (UAS7 ≤6) până în săptămâna 12
- numărul de săptămâni fără angioedem (scorul săptămânal al activităţii angioedemului [AAS7] = 0) până în săptămâna 12.
Răspuns clinic
Atât în cazul REMIX-1, cât și în cel al REMIX-2, criteriile finale principale și toate criteriile secundare au fost îndeplinite și au înregistrat îmbunătățiri semnificative statistic și clinic a simptomelor de prurit și papule la pacienții tratați cu remibrutinib, comparativ cu pacienții cărora li s-a administrat placebo. Rezultatele sunt prezentate în Tabelul 2 și Figura 1.
Tabelul 2 Rezultate privind eficacitatea în REMIX-1 și REMIX-2 în săptămâna 12a,b
| REMIX-1 | REMIX-2 | |||
| Remibrutinib (N=309) | Placebo (N=153) | Remibrutinib (N=297) | Placebo (N=153) | |
| Modificare față de valoarea inițială a UAS7 în săptămâna 12 | ||||
|---|---|---|---|---|
| CFB media LS (ES) | -20,02 (0,716) | -13,79 (0,980) | -19,41 (0,702) | -11,73 (0,948) |
| Diferență CFB media LS (ES) față de placebo | -6,22 (1,136) | -7,68 (1,136) | ||
| IÎ 95% pentru diferență | -8,45, -4,00 | -9,91, -5,46 | ||
| valoare p | <0,001 | <0,001 | ||
| Modificare față de valoarea inițială a ISS7 în săptămâna 12 | ||||
| CFB media LS (ES) | -9,52 (0,343) | -6,89 (0,470) | -8,95 (0,335) | -5,72 (0,454) |
| Diferență CFB media LS (ES) față de placebo | -2,63 (0,544) | -3,23 (0,545) | ||
| IÎ 95% pentru diferență | -3,70, -1,56 | -4,29, -2,16 | ||
| valoare p | <0,001 | <0,001 | ||
| Modificare față de valoarea inițială a HSS7 în săptămâna 12 | ||||
| CFB media LS (ES) | -10,47 (0,401) | -6,86 (0,548) | -10,47 (0,394) | -6,00 (0,531) |
| Diferență CFB media LS (ES) față de placebo | -3,61 (0,635) | -4,47 (0,634) | ||
| IÎ 95% pentru diferență | -4,85, -2,36 | -5,71, -3,23 | ||
| valoare p | <0,001 | <0,001 | ||
| Procent de pacienți cu UAS7 ≤6 în săptămâna 2 | ||||
| n (%) | 104 (33,7) | 5 (3,3) | 89 (30,0) | 9 (5,9) |
| Diferență tratament față de placebo | 30,20 | 24,55 | ||
| (IÎ 95%) | 24,30, 36,10 | 18,31, 30,80 | ||
| valoare p | <0,001 | <0,001 | ||
| Procent de pacienți cu UAS7 ≤6 în săptămâna 12 | ||||
| n (%) | 154 (49,8) | 38 (24,8) | 139 (46,8) | 30 (19,6) |
| Diferență tratament față de placebo | 25,44 | 27,61 | ||
| (IÎ 95%) | 16,48, 34,39 | 19,14, 36,08 | ||
| valoare p | <0,001 | <0,001 | ||
| Procent de pacienți cu UAS7 = 0 în săptămâna 12 | ||||
| n (%) | 96 (31,1) | 16 (10,5) | 83 (27,9) | 10 (6,5) |
| Diferență tratament față de placebo | 20,55 | 21,60 | ||
| (IÎ 95%) | 13,35, 27,75 | 15,10, 28,10 | ||
| valoare p | <0,001 | <0,001 | ||
| Procent de pacienți cu răspuns DLQI = 0-1 în săptămâna 12 | ||||
| n (%) | 120 (39,0) | 34 (22,2) | 106 (35,7) | 28 (18,3) |
| Diferență tratament față de placebo | 17,65 | 18,21 | ||
| (IÎ 95%) | 9,14, 26,16 | 9,96, 26,45 | ||
| valoare p | <0,001 | <0,001 | ||
| Număr cumulat de săptămâni cu UAS7 ≤6 între momentul inițial și săptămâna 12 | ||||
| Media LS (ES) | 5,17 (0,414) | 1,92 (0,241) | 4,50 (0,464) | 1,38 (0,216) |
| Raport rată | 2,69 | 3,26 | ||
| (IÎ 95%) | (2,01, 3,61) | (2,26, 4,71) | ||
| valoare p | <0,001 | <0,001 | ||
| Număr cumulat de săptămâni cu AAS7 = 0 între momentul inițial și săptămâna 12 | ||||
| Media LS (ES) | 8,43 (0,274) | 6,72 (0,330) | 8,81 (0,308) | 6,68 (0,343) |
| Raport rată | 1,25 | 1,32 | ||
| (IÎ 95%) | (1,12, 1,41) | (1,17, 1,49) | ||
| valoare p | <0,001 | <0,001 | ||
| Media LS: media celor mai mici pătrate, ES eroare standard, CFB: modificare față de valoarea inițială, IÎ: interval de încredere, valoare p: valoare p unilaterală, UAS7: scor săptămânal privind activitatea urticariei, scor ISS7: scor săptămânal privind severitatea pruritului, HSS7: scor săptămânal privind severitatea erupțiilor, DLQI: dermatology life quality index, AAS7: scor săptămânal privind activitatea angioedemului. a Toate criteriile finale cu p unilateral nominal p<0,001 b Un criteriu final de la săptămâna 2 (toate celelalte criterii finale sunt de la săptămâna 12) | ||||
Figure 1 Modificare medie față de valoarea inițială a UAS7 până în săptămâna 12 în REMIX-1 și REMIX-2 (date observate)
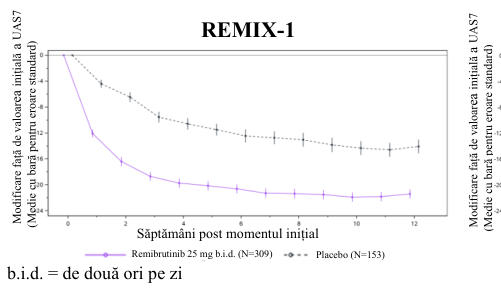
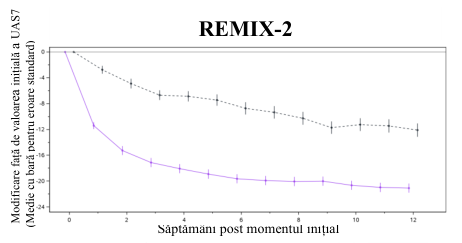
Remibrutinib 25 mg b.i.d. (N=297) Placebo (N=153)
Analizele subgrupelor au demonstrat un beneficiu constant al tratamentului cu remibrutinib față de placebo în cadrul subgrupelor, inclusiv expunere anterioară la medicamente biologice anti-IgE și nivel total de IgE.
Copii și adolescenți
Agenția Europeană pentru Medicamente a suspendat temporar obligația de depunere a rezultatelor studiilor efectuate cu Rhapsido la una sau mai multe subgrupe de copii și adolescenți în UCS (vezi pct. 4.2 pentru informații privind utilizarea la copii și adolescenți).
5.2 Proprietăți farmacocinetice
Absorbție
Remibrutinib este absorbit rapid și atinge Cmax în sânge în jur de aproximativ 1 oră după administrarea dozei în toate dozele studiate (0,5 mg până la 600 mg). Absorbția este considerată a fi, în mare parte, completă (86,9%). Biodisponibilitatea orală absolută este de 33,8%.
Efectul alimentelor
ASC a remibrutinib a crescut cu 33% și Cmax a scăzut cu 5% la administrarea împreună cu o masă bogată în grăsimi, comparativ cu starea de repaus alimentar după administrarea remibrutinib. Remibrutinib poate fi administrat cu sau fără alimente (vezi pct. 4.2).
Distribuție
Remibrutinib este distribuit rapid în celulele sanguine cu un raport sânge-plasmă de 0,813. Legarea de proteinele plasmatice se ridică la 95,4%, fără dependență de concentrație. Pe baza datelor centralizate din analiza farmacocinetică populațională (PopPK), volumul de distribuție la starea de echilibru a fost de 58 litri (compartiment central) și 1 180 litri (compartiment periferic).
Metabolizare
Remibrutinib este metabolizat, în principal, de CYP3A4, ceea ce duce la formarea a 18 metaboliţi inactivi, toţi prezenți în cantităţi mici în circulaţie. Remibrutinib a fost cel mai abundent compus din sânge (16,7%).
Studii in vitro
Metabolizarea in vitro a CYP este determinată, în principal, de CYP3A4. Datele in vitro au arătat că remibrutinib este un substrat al gp-P.
Eliminare
Remibrutinib are un timp mediu de înjumătățire plasmatică prin eliminare cuprins între 1 și 2 ore la starea de echilibru. Clearance-ul oral aparent mediu la starea de echilibru (Clss/F), determinat prin analiza PopPK, este de 160 litri/h. După administrarea intravenoasă a 100 mg [14C]-remibrutinib, eliminarea radioactivităţii (remibrutinib şi metaboliţi) a fost de aproximativ 72,9% din doza administrată în materiile fecale şi 27,1% în urină. Excreția renală a remibrutinib nemodificat după administrarea orală a fost sub 1% din doză.
Liniaritate/Non-liniaritate
Farmacocinetica remibrutinib la starea de echilibru este aproximativ liniară în intervalul total de doze zilnice de 10 până la 200 mg.
Relație(i) farmacocinetică(e)/farmacodinamică(e)
Datele clinice farmacocinetice şi farmacodinamice (PK/PD) au estimat un grad de ocupare a BTK de ≥96% în sânge, menţinut pe parcursul întregii zile cu remibrutinib 25 mg de două ori pe zi.
Grupe speciale de pacienți
Analiza PopPK a arătat că nu există efecte relevante din punct de vedere clinic ale vârstei (18 până la 80 ani), sexului (63,5% femei și 36,5% bărbați), rasei/etniei (59,3% non-asiatică, 8,8% chineză continentală, 12,2% japoneză și 19,7% alte etnii asiatice) și greutății corporale (39 la 162 kg; medie 74,8 kg) asupra farmacocineticii remibrutinib.
Insuficienţă renală
Efectele insuficienţei renale asupra farmacocineticii remibrutinib nu au fost evaluate într-un studiu clinic dedicat. Într-o analiză PopPK, nu s-a observat nicio relație semnificativă din punct de vedere clinic între testele funcției renale și farmacocinetica remibrutinib. În analiza PopPK, au existat 19,3%, 2,2% și 0,1% dintre subiecții cu insuficiență renală ușoară, moderată, respectiv, severă.
Insuficienţă hepatică
Cmax și ASC ale remibrutinib la starea de echilibru au crescut de 1,85 ori și 2,15 ori la subiecții cu insuficiență hepatică ușoară (clasa A Child-Pugh), de 1,65 ori și 2,07 ori la subiecții cu insuficiență hepatică moderată (clasa B Child-Pugh) și de 1,99 ori, respectiv 3,12 ori la subiecții cu insuficiență hepatică severă (clasa C Child-Pugh), comparativ cu subiecții cu funcție hepatică normală, după administrarea orală a unei doze de remibrutinib 25 mg de două ori pe zi. Nu a existat nicio modificare a legării de proteinele plasmatice a remibrutinib la subiecţii cu insuficienţă hepatică, comparativ cu subiecţii cu funcţie hepatică normală (vezi pct. 4.2).
Copii și adolescenți
Nu au fost efectuate studii farmacocinetice cu remibrutinib la pacienţi cu vârsta sub 18 ani.
5.3 Date preclinice de siguranță
Remibrutinib a inhibat răspunsurile primare ale anticorpilor în studiile farmacologice la rozătoare și a crescut timpul de sângerare la nivelul cozii la șobolan în evaluările hemostazei. Aceste observații, care au apărut la expuneri relevante din punct de vedere farmacologic și clinic, au fost considerate legate de efectele remibrutinib asupra funcțiilor specifice ale celulelor B, respectiv ale trombocitelor. Datele non-clinice nu au evidențiat niciun risc special suplimentar pentru om pe baza studiilor convenționale farmacologice privind evaluarea siguranței, toxicitatea după doze repetate, genotoxicitatea, carcinogenitatea și fototoxicitatea.
Toxicitatea funcției de reproducere
În studiile de dezvoltare embrio-fetală (EFD) la femele gestante de iepure, au crescut malformaţiile externe fetale (ochi deschişi/opaci, fălci mici, hiperflexia membrelor anterioare) şi toxicitatea maternă (consum redus, tranzitoriu, de alimente şi semne clinice adverse) au apărut la o doză de aproximativ 141 ori mai mare decât doza maximă recomandată la om (MRHD) de 25 mg, de două ori pe zi, fără a se observa o marjă de siguranţă bazată pe efecte adverse (NOAEL) de 23 ori mai mare decât MRHD de 25 mg, de două ori pe zi, pe baza ASC. S-a considerat că este puţin probabil ca rezultatele fetale să fie secundare toxicităţii materne. Nu s-a observat niciun efect asupra EFD la șobolan, cu o marjă de siguranță bazată pe NOAEL de 126 ori mai mare în ceea ce privește ASC la starea de echilibru comparativ cu expunerea la om la MRHD.
Într-un studiu privind dezvoltarea prenatală și postnatală (PPND) la șobolan, remibrutinib a indus efecte adverse care au afectat femele (stare muribundă și semne clinice de toxicitate, durate ale gestației ușor crescute) și descendenții până în ziua 1 de lactație (număr mediu ușor crescut de pui morți sau absenți și număr mediu mai mic al puilor născuți de o femelă la aceeași fătare) cu o marjă de siguranță bazată pe NOAEL pentru femele și descendenți de aproximativ 67 ori mai mare decât MRHD de 25 mg, de două ori pe zi, pe baza ASC. Nu s-au observat efecte adverse pentru descendenții supraviețuitori care s-au dezvoltat până la vârsta adultă.
Într-un studiu privind fertilitatea la șobolan, remibrutinib nu a avut un impact asupra fertilității la șobolan, femele sau masculi, până la expuneri maxime realizabile de 79 și 15 ori mai mari decât MRHD de 25 mg de două ori pe zi, pe baza ASC.
6. PROPRIETĂȚI FARMACEUTICE
6.1 Lista excipienților
Nucleul comprimatului
Manitol
Celuloză microcristalină
Copovidonă
Croscarmeloză sodică
Fumarat stearil sodic
Lauril sulfat de sodiu
Învelișul comprimatului
Alcool polivinilic
Macrogol 4000
Talc
Dioxid de titan (E171)
Oxid galben de fier (E172)
Oxid roșu de fier (E172)
6.2 Incompatibilități
Nu este cazul.
6.3 Perioada de valabilitate
2 ani
6.4 Precauții speciale pentru păstrare
A se păstra în ambalajul original pentru a fi protejat de umiditate.
6.5 Natura și conținutul ambalajului
Rhapsido este disponibil în blistere PA/Al/PVC/Al (poliamidă/aluminiu/policlorură de vinil/aluminiu) cu folie de susținere din aluminiu, în ambalaje care conțin 30, 60 sau 180 comprimate filmate.
Este posibil ca nu toate mărimile de ambalaj să fie comercializate.
6.6 Precauții speciale pentru eliminarea reziduurilor
Orice medicament neutilizat sau material rezidual trebuie eliminat în conformitate cu reglementările locale.
7. DEȚINĂTORUL AUTORIZAȚIEI DE PUNERE PE PIAȚĂ
Novartis Europharm Limited
Vista Building
Elm Park, Merrion Road
Dublin 4
Irlanda
8. NUMĂRUL(ELE) AUTORIZAȚIEI DE PUNERE PE PIAȚĂ
EU/1/26/2024/001-003
9. DATA PRIMEI AUTORIZĂRI SAU A REÎNNOIRII AUTORIZAȚIEI
10. DATA REVIZUIRII TEXTULUI
Informații detaliate privind acest medicament sunt disponibile pe site-ul Agenției Europene pentru Medicamente https://www.ema.europa.eu.