DECITABINA TEVA 50 mg
Rezumatul caracteristicilor produsului (RCP)
- 1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI
- 2. COMPOZIŢIA CALITATIVĂ ŞI CANTITATIVĂ
- 3. FORMA FARMACEUTICĂ
-
4. DATE CLINICE
- 4.1 Indicaţii terapeutice
- 4.2 Doze şi mod de administrare
- 4.3 Contraindicaţii
- 4.4 Atenţionări şi precauţii speciale pentru utilizare
- 4.5 Interacţiuni cu alte medicamente şi alte forme de interacţiune
- 4.6 Fertilitatea, sarcina şi alăptarea
- 4.7 Efecte asupra capacităţii de a conduce vehicule şi de a folosi utilaje
- 4.8 Reacţii adverse
- 4.9 Supradozaj
- 5. PROPRIETĂŢI FARMACOLOGICE
- 6. PROPRIETĂŢI FARMACEUTICE
- 7. DEŢINĂTORUL AUTORIZAŢIEI DE PUNERE PE PIAŢĂ
- 8. NUMĂRUL(ELE) AUTORIZAŢIEI DE PUNERE PE PIAŢĂ
- 9. DATA PRIMEI AUTORIZĂRI SAU A REÎNNOIRII AUTORIZAŢIEI
- 10. DATA REVIZUIRII TEXTULUI
1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI
Decitabină Teva 50 mg pulbere pentru concentrat pentru soluţie perfuzabilă
2. COMPOZIŢIA CALITATIVĂ ŞI CANTITATIVĂ
Un flacon de pulbere pentru concentrat pentru soluţie perfuzabilă conţine decitabină 50 mg.
După reconstituire cu 10 ml de apă pentru preparate injectabile, fiecare ml de concentrat conţine 5 mg de decitabină.
Pentru lista tuturor excipienţilor, a se vedea pct. 6.1.
3. FORMA FARMACEUTICĂ
Pulbere pentru concentrat pentru soluţie perfuzabilă (pulbere pentru perfuzie).
Pulbere liofilizată de culoare albă până la aproape albă.
4. DATE CLINICE
4.1 Indicaţii terapeutice
Decitabină Teva este indicat în tratamentul pacienţilor adulţi, nou diagnosticaţi cu leucemie mieloidă acută (LMA) de novo sau secundară, în conformitate cu clasificarea Organizaţiei Mondiale a Sănătăţii (OMS) care nu sunt eligibili pentru chimioterapie standard de inducţie.
4.2 Doze şi mod de administrare
Administrarea Decitabină Teva trebuie iniţiată sub supravegherea unor medici cu experienţă în utilizarea medicamentelor pentru chimioterapie.
Doze
Într-un ciclu de tratament, Decitabină Teva se administrează în doză de 20 mg/m2 suprafaţă corporală, prin perfuzie intravenoasă cu durata de 1 oră, cu repetare zilnică timp de 5 zile consecutive (de exemplu, un total de 5 doze per ciclu de tratament). Doza zilnică totală nu trebuie să depăşească 20 mg/m2, iar doza totală per ciclu de tratament nu trebuie să depăşească 100 mg/m2. În cazul omiterii unei doze, tratamentul trebuie reluat cât mai repede posibil. Ciclul trebuie repetat o dată la 4 săptămâni, în funcţie de răspunsul clinic al pacientului şi de toxicitatea observată. Se recomandă ca pacienţii să urmeze minimum 4 cicluri de tratament; cu toate acestea, pentru obţinerea unei remisiuni complete sau parţiale pot fi necesare mai mult de 4 cicluri. Tratamentul poate fi continuat atâta timp cât pacientul are un răspuns, continuă să beneficieze sau prezintă boală stabilă, de exemplu, în absenţa progresiei evidente.
În cazul în care după 4 cicluri de tratament, valorile hematologice ale pacientului (de exemplu, numărul de trombocite sau numărul absolut de neutrofile), nu revin la valori preterapeutice sau dacă apare progresia bolii (numărul celulelor blastice periferice este în creştere sau valorile celulelor blastice medulare se deteriorează), se poate considera că pacientul nu răspunde la tratament şi trebuie avute în vedere opţiuni terapeutice alternative la Decitabină Teva.
Nu se recomandă administrarea de rutină, înainte de tratament a pre-medicaţiei pentru prevenirea senzaţiei de greaţă şi vărsăturilor, dar se poate administra dacă este cazul.
Managementul mielosupresiei şi al complicaţiilor asociate
Mielosupresia şi reacţiile adverse corelate cu mielosupresia (trombocitopenia, anemia, neutropenia şi neutropenia febrilă) sunt frecvente la pacienţii cu LMA indiferent dacă primesc sau nu tratament. Complicaţiile mielosupresiei includ infecţii şi sângerări.Tratamentul poate fi amânat, la latitudinea medicului curant, în cazul în care pacientul prezintă complicaţii asociate mielosupresiei, cum sunt cele descrise mai jos:
- Neutropenie febrilă (temperatură ≥ 38,5°C şi număr absolut de neutrofile < 1000/μl)
- Infecţii virale, bacteriene sau fungice active (de exemplu, necesită administrarea intravenoasă de antiinfecţioase sau de tratament suportiv extensiv)
- Hemoragie (la nivel gastrointestinal, genito-urinar, pulmonar însoţită de valori ale trombocitelor < 25000/μl sau orice hemoragie la nivelul sistemului nervos central)
Tratamentul cu Decitabină Teva poate fi reluat după ce aceste afecţiuni au fost ameliorate sau au fost stabilizate prin tratament adecvat (terapie antiinfecţioasă, transfuzii sau factori de creştere).
În studiile clinice, aproximativ o treime dintre pacienţii în tratament cu Decitabină Teva au necesitat o întârziere a dozei. Nu este recomandată reducerea dozei.
Copii şi adolescenţi
Decitabină Teva nu trebuie utilizat la copiii cu LMA cu vârsta < 18 ani, deoarece eficacitatea nu a fost stabilită. Datele disponibile în prezent sunt descrise la pct. 4.8, 5.1 şi 5.2.
Insuficienţă hepatică
Nu au fost efectuate studii la pacienţi cu insuficienţă hepatică. Nu a fost evaluată necesitatea ajustării dozelor la pacienţii cu insuficienţă hepatică. Dacă se produce agravarea funcţiei hepatice, pacienţii trebuie monitorizaţi cu atenţie (a se vedea pct. 4.4 şi 5.2).
Insuficienţă renală
Nu au fost efectuate studii la pacienţi cu insuficienţă renală. Nu a fost evaluată necesitatea ajustării dozelor la pacienţii cu insuficienţă renală (a se vedea pct. 4.4 şi 5.2).
Mod de administrare
Decitabină Teva se administrează prin perfuzie intravenoasă. Nu este necesar un cateter venos central.
Pentru instrucţiuni privind reconstituirea şi diluarea medicamentului înainte de administrare, a se vedea pct. 6.6.
4.3 Contraindicaţii
Hipersensibilitate la decitabină sau la oricare dintre excipienţii enumeraţi la pct. 6.1.
Alăptare (a se vedea pct.4.6).
4.4 Atenţionări şi precauţii speciale pentru utilizare
Mielosupresia
Mielosupresia şi complicaţiile asociate mielosupresiei, inclusiv infecţiile şi sângerările care apar la pacienţii cu LMA se pot exacerba în timpul tratamentului cu Decitabină Teva. Prin urmare, pacienţii prezintă un risc crescut de infecţii severe (cauzate de agenţi patogeni bacterieni, fungici şi virali), cu potenţial letal (a se vedea pct. 4.8). Pacienţii trebuie monitorizaţi pentru semne şi simptome de infecţii şi trataţi imediat.
În studiile clinice, majoritatea pacienţilor au prezentat iniţial mielosupresie de grad 3/4. La pacienţii care au prezentat iniţial anomalii de gradul 2, agravarea mielosupresiei a fost observată la majoritatea pacienţilor şi mai frecvent comparativ cu pacienţii care iniţial au prezentat anomalii de grad 1 sau 0. Mielosupresia cauzată de Decitabină Teva este reversibilă. Trebuie efectuate în mod regulat hemoleucograma completă şi numărul de trombocite, aşa cum este indicat clinic şi înainte de fiecare ciclu de tratament. În prezenţa mielosupresiei sau a complicaţiilor sale, tratamentul cu Decitabină Teva poate fi întrerupt şi/sau se pot institui măsuri suportive (a se vedea pct. 4.2 şi 4.8).
Tulburări respiratorii, toracice și mediastinale
La pacienții tratați cu decitabină au fost raportate cazuri de boală pulmonară interstițială (BPI) (inclusiv infiltrate pulmonare, pneumonie organizată și fibroză pulmonară), fără semne de etiologie infecțioasă. Pacienții cu debut acut sau cu agravare inexplicabilă a simptomelor pulmonare trebuie evaluați cu atenție pentru a exclude BPI. În cazul în care se confirmă BPI, trebuie inițiat tratamentul adecvat (a se vedea pct 4.8).
Insuficienţă hepatică
Utilizarea la pacienţii cu insuficienţă hepatică nu a fost stabilită. Se recomandă precauţie în administrarea Decitabină Teva la pacienţi cu insuficienţă hepatică şi la pacienții care dezvoltă semne și simptome de insuficiență hepatică. Testele funcției hepatice trebuie efectuate înainte de inițierea tratamentului și înainte de fiecare ciclu de tratament, și după cum este indicat din punct de vedere clinic (a se vedea pct. 4.2 şi 5.2).
Insuficienţă renală
Utilizarea la pacienţii cu insuficienţă renală severă nu a fost studiată. Se recomandă precauţie în administrarea Decitabină Teva la pacienţi cu insuficienţă renală severă (clearance al creatininei [ClCr] < 30 ml/min). Testele funcției renale trebuie efectuate înainte de inițierea terapiei și înainte de fiecare ciclu de tratament, și după cum este indicat din punct de vedere clinic (a se vedea pct. 4.2).
Boală cardiacă
Pacienţii cu antecedente de insuficienţă cardiacă congestivă severă sau boală cardiacă instabilă clinic au fost excluşi din studiile clinice şi, prin urmare, nu a fost stabilită siguranţa şi eficacitatea Decitabină Teva la aceşti pacienţi. După punerea pe piaţă a medicamentului, au fost raportate cazuri de cardiomiopatie cu decompensare cardiacă, în unele cazuri reversibilă după întreruperea tratamentului, reducerea dozei sau tratament corectiv. Pacienţii trebuie monitorizaţi pentru depistarea semnelor şi a simptomelor de insuficienţă cardiacă, în special cei cu antecedente de boală cardiacă.
Sindrom de diferențiere
Au fost raportate cazuri de sindrom de diferențiere (cunoscut și sub numele de sindromul acidului retinoic) la pacienții cărora li s-a administrat decitabină. Sindromul de diferențiere poate fi letal (a se vedea pct. 4.8). Tratamentul cu corticosteroizi IV în doze mari și monitorizarea hemodinamică trebuie luate în considerare la debutul simptomelor sau semnelor sugestive ale sindromului de diferențiere. Trebuie luată în considerare întreruperea temporară a tratamentului cu Decitabină Teva până la dispariția simptomelor și dacă tratamentul este reluat, se recomandă prudență.
Excipienţi
Acest medicament conține mai puțin de 1 mmol potasiu (39 mg) per flacon, adică este practic ‘fără potasiu’.
Acest medicament conține mai puțin de 1 mmol sodiu (23 mg) per flacon, adică este practic ‘fără sodiu.
4.5 Interacţiuni cu alte medicamente şi alte forme de interacţiune
Nu s-au efectuat studii clinice specifice privind interacţiunile medicamentoase cu decitabina.
Există potenţialul de interacţiune cu alte medicamente care sunt activate de asemenea prin fosforilare secvenţială (prin intermediul activităţilor fosfokinazei intracelulare) şi/sau metabolizate de către enzimele implicate în inactivarea decitabinei (de exemplu, citidin deaminaza). Prin urmare, trebuie manifestată precauţie, dacă aceste substanțe active sunt administrate în asociere cu decitabina.
Impactul medicamentelor administrate concomitent asupra decitabinei
Nu se anticipează interacţiuni metabolice mediate de citocromul CYP 450, deoarece metabolizarea decitabinei nu este mediată de către acest sistem, ci prin dezaminare oxidativă.
Impactul decitabinei asupra medicamentelor administrate concomitent
Având în vedere faptul că, in vitro legarea la proteinele transportatoare plasmatice este scăzută (< 1%), este puţin probabil ca decitabina să înlocuiască medicamentele administrate concomitent, de la nivelul proteinelor transportoare ale acestora. In vitro, s-a demonstrat că decitabina este un inhibitor slab al transportului mediat de gp P şi, prin urmare, nu este anticipată afectarea transportului mediat de gp P al medicamentelor administrate concomitent (a se vedea pct. 5.2).
4.6 Fertilitatea, sarcina şi alăptarea
Femei aflate la vârsta fertilă/Contracepţia la bărbaţi şi femei
Din cauza potențialului genotoxic al decitabinei (a se vedea pct. 5.3), femeile aflate la vârsta fertilă trebuie să utilizeze măsuri contraceptive eficace şi trebuie să evite să rămână gravide în timpul tratamentului cu Decitabină Teva și timp de 6 luni după încheierea tratamentului. Bărbaţii trebuie să utilizeze măsuri contraceptive eficace şi să fie sfătuiţi să nu conceapă un copil în timpul tratamentului cu Decitabină Teva şi timp de 3 luni după încheierea tratamentului (a se vedea pct. 5.3).
Nu a fost studiată utilizarea decitabinei în asociere cu contraceptive hormonale.
Sarcina
Nu există date adecvate cu privire la utilizarea Decitabină Teva la femeile gravide. Studiile au arătat că decitabina este teratogenă la şobolani şi şoareci (a se vedea pct. 5.3). Nu este cunoscut riscul potenţial pentru om. Pe baza rezultatelor studiilor la animale şi a mecanismului de acţiune, Decitabină Teva nu trebuie utilizat în timpul sarcinii şi la femeile aflate la vârsta fertilă care nu folosesc metode de contracepţie eficace. Înainte de începerea tratamentului, trebuie efectuat un test de sarcină la toate femeile aflate la vârsta fertilă. Pacienta trebuie informată cu privire la riscul potenţial pentru făt în cazul în care Decitabină Teva este utilizat în timpul sarcinii sau în cazul în care pacienta rămâne gravidă în timpul tratamentului cu acest medicament.
Alăptarea
Nu este cunoscut dacă decitabina sau metaboliţii săi se excretă în laptele matern. Decitabină Teva este contraindicat în timpul alăptării; prin urmare, dacă tratamentul cu acest medicament este necesar, alăptarea trebuie întreruptă (a se vedea pct. 4.3).
Fertilitatea
Nu sunt disponibile date privind efectul decitabinei asupra fertilităţii. În studiile non-clinice la animale, decitabina modifică fertilitatea masculină şi are efecte mutagene. Datorită posibilităţii de infertilitate, ca o consecinţă a tratamentului cu Decitabină Teva, bărbaţii trebuie să solicite consiliere privind conservarea spermei, iar pacientele de sex feminin aflate în perioada fertilă trebuie să solicite consult cu privire la crioconservarea ovocitului, înainte de iniţierea tratamentului.
4.7 Efecte asupra capacităţii de a conduce vehicule şi de a folosi utilaje
Decitabină Teva are influenţă moderată asupra capacităţii de a conduce vehicule și de a folosi utilaje. Pacienţii trebuie informaţi că pot avea reacţii adverse cum este anemia în timpul tratamentului. Prin urmare, se recomandă prudenţă la conducerea vehiculelor sau folosirea utilajelor.
4.8 Reacţii adverse
Rezumatul profilului de siguranţă
Reacţiile adverse la medicament raportate cel mai frecvent (≥ 35%) sunt febra, anemia şi trombocitopenia.
Cele mai frecvente reacţii adverse de grad 3/4 (≥ 20%) la medicament au inclus pneumonia, trombocitopenia, neutropenia, neutropenia febrilă şi anemia.
În studiile clinice, 30% dintre pacienţi trataţi cu decitabină şi 25% dintre pacienţii trataţi în braţul comparator au avut evenimente adverse care au determinat deces în timpul tratamentului sau în perioada de 30 de zile după administrarea ultimei doze din medicaţia de studiu.
În grupul de tratament cu decitabină, a fost o incidenţă mai mare a întreruperii tratamentului din cauza evenimentelor adverse la femei, comparativ cu bărbaţi (43% comparativ cu 32%).
Lista reacţiilor adverse la medicament în format tabelar
Reacţiile adverse la medicament raportate la 293 pacienţi cu LMA trataţi cu decitabină sunt rezumate în Tabelul 1. Tabelul următor prezintă date din studiile clinice pentru LMA şi din experienţa de după punerea pe piaţă. Reacţiile adverse la medicament sunt prezentate în funcţie de categoria de frecvenţă. Categoriile de frecvenţă sunt definite după cum urmează: foarte frecvente (≥ 1/10), frecvente (≥ 1/100 şi < 1/10), mai puţin frecvente (≥ 1/1000 şi < 1/100), rare (≥ 1/10000 şi < 1/1000), foarte rare (< 1/10000), cu frecvenţă necunoscută (care nu poate fi estimată din datele disponibile).
În cadrul fiecărei grupe de frecvenţă, reacţiile adverse la medicament sunt prezentate în ordine descrescătoare a gravităţii.
Tabelul 1: Reacţiile adverse la medicament identificate cu decitabină
| Aparate, sisteme şi organe | Frecvenţa (toate gradele) | Reacţie adversă la medicament | Frecvenţa | |
| Toate gradele (%) | Gradele 3-4a (%) | |||
| Infecţii şi infestări | Foarte frecvente | pneumonie* | 24 | 20 |
| infecţie la nivelul tractului urinar* | 15 | 7 | ||
| orice alte infecţii (virale, bacteriene şi fungice)*,b,c,d | 63 | 39 | ||
| Frecvente | şoc septic* | 6 | 4 | |
| sepsis* | 9 | 8 | ||
| sinuzită | 3 | 1 | ||
| Neoplasme benigne, maligne și nespecificate (inclusiv chisturi și polipi) | Cu frecvență necunoscută | sindrom de diferențiere | necunoscută | necunoscută |
| Tulburări hematologice şi limfatice | Foarte frecvente | neutropenie febrilă* | 34 | 32 |
| neutropenie* | 32 | 30 | ||
| trombocitopenie*, e | 41 | 38 | ||
| anemie | 38 | 31 | ||
| leucopenie | 20 | 18 | ||
| Mai puţin frecvente | pancitopenie* | < 1 | < 1 | |
| Tulburări ale sistemului imunitar | Frecvente | hipersensibilitate inclusiv reacţie anafilacticăf | 1 | < 1 |
| Tulburări metabolice și de nutriție | Foarte frecvente | hiperglicemie | 13 | 3 |
| Tulburări ale sistemului nervos | Foarte frecvente | cefalee | 16 | 1 |
| Tulburări cardiace | Mai puţin frecvente | cardiomiopatie | < 1 | < 1 |
| Tulburări respiratorii, toracice şi mediastinale | Foarte frecvente | epistaxis | 14 | 2 |
| Cu frecvență necunoscută | boală pulmonară interstițială | necunoscută | necunoscută | |
| Tulburări gastrointestinale | Foarte frecvente | diaree | 31 | 2 |
| vărsături | 18 | 1 | ||
| greaţă | 33 | < 1 | ||
| Frecvente | stomatită | 7 | 1 | |
| Cu frecvenţă necunoscută | enterocolită, inclusiv colită neutropenică, cecită* | necunoscută | necunoscută | |
| Tulburări hepatobiliare | Foarte frecvente | disfuncție hepatică | 11 | 3 |
| Frecvente | hiperbilirubinemieg | 5 | < 1 | |
| Afecţiuni cutanate şi ale ţesutului subcutanat | Mai puţin frecvente | dermatoză acută febrilă neutrofilică (sindromul Sweet) | < 1 | NA |
| Tulburări generale şi la nivelul locului de administrare | Foarte frecvente | febră | 48 | 9 |
a Criteriile de Terminologie Comună privind gradul evenimentelor adverse cel mai grave ale Institutului Naţional al Cancerului
b Excluzând pneumonia, infecţiile de tract urinar, sepsis, şoc septic şi sinuzită
c „Celelalte infecţii” cel mai frecvent raportate în studiul DACO-016 au fost: herpes oral, candidoză orală, faringită, infecţii ale căilor respiratorii superioare, celulită, bronşită, nazofaringită
d inclusiv enterocolită infecţioasă
e Inclusiv hemoragiile asociate cu trombocitopenie, inclusiv cazurile letale
f Inclusiv termeni preferaţi hipersensibilitate, hipersensibilitate la medicament, reacţie anafilactică, şoc anafilactic, reacţie anafilactoidă, şoc anafilactoid.
g În studiile clinice privind LMA și sindromul mielodisplazic (SMD), frecvența de raportare a hiperbilirubinemiei a fost de 11% pentru Toate gradele și de 2% pentru Gradele 3-4.
* Include evenimentele cu un rezultat letal.
N/A = Nu se aplică
Descrierea reacţiilor adverse selectate
Reacţii adverse hematologice
Reacţiile adverse hematologice la medicament, cel mai frecvent raportate în în timpul tratamentului cu Decitabină Teva au inclus neutropenie febrilă, trombocitopenie, neutropenie, anemie şi leucopenie.
La pacienţii trataţi cu decitabină s-au raportat reacţii adverse grave la medicament asociate hemoragiei, unele dintre acestea, în contextul trombocitopeniei severe, având ca rezultat decesul, precum hemoragie la nivelul sistemului nervos central (SNC) (2%) şi hemoragie gastrointestinală (GI) (2%).
Reacţiile adverse hematologice trebuie gestionate prin monitorizarea de rutină a hemoleucogramei şi iniţierea precoce a tratamentelor de susţinere, în funcţie de necesităţi. Tratamentele de susţinere includ administrarea profilactică de antibiotice şi/sau administrarea factorului de creştere (de exemplu, GCSF) în caz de neutropenie şi transfuzii sanguine pentru anemie sau trombocitopenie, în funcţie de recomandările din ghidurile de diagnostic și tratament naționale. Pentru situaţiile în care administrarea decitabinei trebuie amânată, a se vedea pct. 4.2.
Reacţii adverse la medicament infecţii şi infestări
La pacienţii trataţi cu decitabină, s-au raportat reacţii adverse grave la medicament asociate unei infecţii, cu potenţial letal, cum sunt şoc septic, sepsis, pneumonie şi alte infecţii (virale, bacteriene şi fungice).
Tulburări gastro-intestinale
În timpul tratamentului cu decitabină au fost raportate cazuri de enterocolită, care includ colită neutropenică şi cecită. Enterocolita poate duce la complicaţii septice şi se poate asocia cu rezultate letale.
Tulburări respiratorii, toracice și mediastinale
La pacienții tratați cu decitabină au fost raportate cazuri de boală pulmonară interstițială (care includ infiltrate pulmonare, pneumonie organizată și fibroză pulmonară), fără semne de etiologie infecțioasă.
Sindrom de diferențiere
Au fost raportate cazuri de sindrom de diferențiere (cunoscut și sub numele de sindromul acidului retinoic) la pacienții cărora li s-a administrat decitabină. Sindromul de diferențiere poate fi letal iar simptomele și manifestările clinice includ insuficiență respiratorie, infiltrate pulmonare, febră, erupție cutanată, edem pulmonar, edem periferic, creștere rapidă în greutate, revărsate pleurale, revărsate pericardice, hipotensiune arterială și disfuncție renală. Sindromul de diferențiere poate apărea cu sau fără leucocitoză concomitentă. De asemenea, poate apărea sindrom de scurgere capilară și coagulopatie (a se vedea pct. 4.4).
Copii şi adolescenţi
Evaluarea siguranţei la pacienţii copii şi adolescenţi se bazează pe date limitate privind siguranţa, obţinute în cadrul unui studiu de fază I/II de evaluare a farmacocineticii, siguranţei şi eficacităţii decitabinei la copii şi adolescenţi (cu vârsta între 1 şi 14 ani) cu LMA recidivantă sau refractară la tratament (n = 17) (a se vedea pct. 5.1). Nu au fost observate probleme legate de siguranţă în cadrul acestui studiu efectuat la copii şi adolescenţi.
Raportarea reacţiilor adverse suspectate
Raportarea reacţiilor adverse suspectate după autorizarea medicamentului este importantă. Acest lucru permite monitorizarea continuă a raportului beneficiu/risc al medicamentului. Profesioniştii din domeniul sănătăţii sunt rugaţi să raporteze orice reacţie adversă suspectată la
Agenţia Naţională a Medicamentului şi a Dispozitivelor Medicale din România
Str. Aviator Sănătescu nr. 48, sector 1
Bucureşti 011478- RO e-mail: adr@anm.ro
Website: www.anm.ro
4.9 Supradozaj
Nu există experienţă directă privind supradozajul la om şi nici antidot specific. Cu toate acestea, datele din primele studii clinice publicate în literatura de specialitate menţionează mielosupresie crescută, care include neutropenie prelungită şi trombocitopenie prelungită la doze de 20 de ori mai mari decât doza terapeutică obișnuită. Toxicitatea este probabil să se manifeste ca exacerbare a reacţiilor adverse la medicament, în principal mielosupresie. Tratamentul în caz de supradozaj trebuie să fie suportiv.
5. PROPRIETĂŢI FARMACOLOGICE
5.1 Proprietăţi farmacodinamice
Grupa farmacoterapeutică: Agenţi antineoplazici, antimetaboliţi, analogi ai pirimidinei;
Cod ATC: L01BC08
Mecanismul de acţiune
Decitabina (5-aza-2’-deoxicitidina) este un analog al deoxinucleozid citidinei care inhibă selectiv ADN metiltransferaza la doze mici, având ca rezultat hipometilarea în promotorul genei, care poate conduce la reactivarea genelor de supresie tumorală, inducerea diferenţierii sau senescenţei celulare urmată de apoptoză.
Experienţa clinică
Utilizarea decitabină a fost studiată într-un studiu de fază III, deschis, randomizat, multicentric, (DACO-016) la subiecţii nou diagnosticaţi cu LMA de novo sau secundară, în conformitate cu clasificarea OMS. Decitabina (n = 242) a fost comparat cu opţiunea de tratament (OT, n = 243), care a constat în alegerea pacientului împreună cu recomandarea medicului de a primi fie numai îngrijiri de susţinere (n = 28, 11,5%) sau citarabină 20 mg/m2 administrată subcutanat o dată pe zi, timp de 10 zile consecutive, repetată la fiecare 4 săptămâni (n = 215, 88,5%). Decitabina a fost administrat ca perfuzie intravenoasă cu durata de 1 oră, în doză de 20 mg/m2 o dată pe zi, timp de 5 zile consecutive, în administrare repetată la fiecare 4 săptămâni.
Subiecţii care au fost consideraţi candidaţi pentru chimioterapia standard de inducţie nu au fost incluşi în studiu, după cum arată caracteristicile iniţiale următoare. Vârsta mediană pentru populaţia cu intenţie de tratament (ITT) a fost de 73 de ani (interval cuprins între 64 şi 91 de ani). Treizeci şi şase la sută dintre subiecţi aveau risc citogenetic scăzut la momentul iniţial. Restul subiecţilor aveau risc citogenetic intermediar. Pacienţii cu citogenetică favorabilă nu au fost incluşi în studiu. Douăzeci şi cinci la sută dintre subiecţi au avut un status de performanţă ECOG [Eastern Cooperative Oncology Group]≥ 2. Optzeci şi unu la sută dintre subiecţi aveau comorbidităţi semnificative (de exemplu, infecţie, insuficienţă cardiacă, insuficienţă pulmonară). Numărul de pacienţi trataţi cu decitabină în funcţie de grupul rasial a fost: caucazieni 209 (86,4%) şi asiatici 33 (13,6%).
Criteriul final principal de evaluare al studiului a fost supravieţuirea globală. Criteriul final secundar de evaluare a fost rata de remisiune completă, care a fost evaluată de o comisie independentă de experţi. Supravieţuirea în absenţa progresiei bolii şi supravieţuirea fără evenimente au fost criteriile finale terţiare de evaluare.
Supravieţuirea generală mediană, în populaţia cu ITT a fost de 7,7 luni la subiecţii trataţi cu decitabină, comparativ cu 5,0 luni la subiecții din braţul OT (rata riscului 0,85; IÎ 95%: 0,69, 1,04, p = 0,1079). Diferenţa nu a atins semnificaţie statistică, cu toate acestea, a existat o tendinţă de îmbunătăţire a ratei de supravieţuire cu o reducere de 15% a riscului de deces pentru subiecţii din brațul de tratament cu decitabină (Figura 1). Atunci când s-a cenzurat pentru o terapie ulterioară cu potenţial de modificare a bolii (de exemplu, chimioterapie de inducţie sau agent de hipometilare), analiza pentru supravieţuirea globală a arătat o reducere cu 20% a riscului de deces pentru pacienţii din grupul de tratament cu decitabină [RR = 0,80, (IÎ 95%: 0,64, 0,99), valoarea p = 0,0437)].
Figura 1. Supravieţuirea generală (Populaţia ITT)
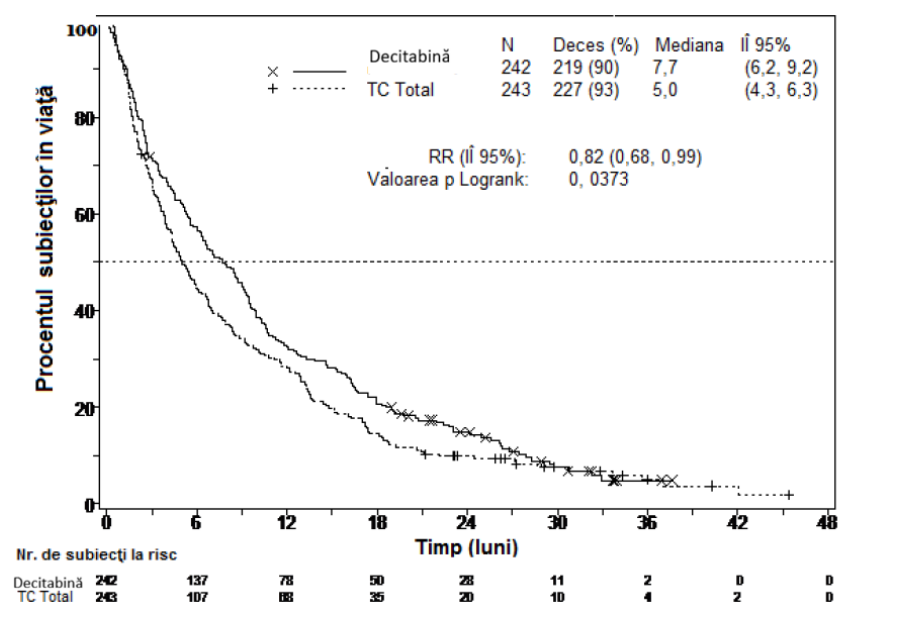
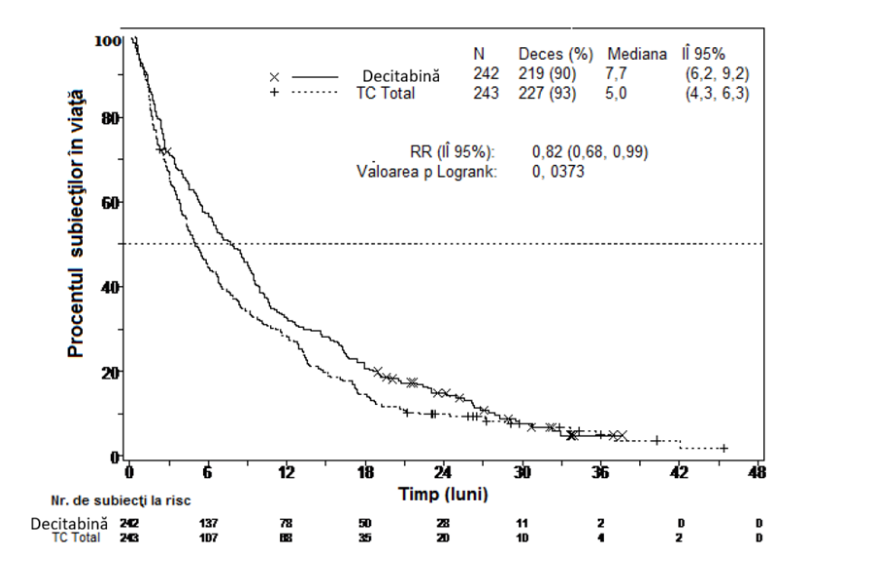
Într-o analiză care a inclus suplimentar 1 an de date de supravieţuire mature, efectul decitabină asupra supravieţuirii globale a demonstrat o îmbunătăţire clinică comparativ cu braţul OT (7,7 luni faţă de 5,0 luni, respectiv, rata de risc = 0,82, IÎ 95%: 0,68, 0,99, valoarea nominală p = 0,0373, Figura 2).
Figura 2. Analiza datelor mature de supravieţuire globală (Populaţia ITT)
Pe baza analizei populaţiei în ITT, s-a obţinut o diferenţă semnificativă statistic a ratei de remisiune completă (RC+RCp) în favoarea subiecţilor din braţul în tratament cu decitabină, 17,8% (43/242), comparativ cu braţul OT, 7,8% (19/243); diferenţa de tratament de 9,9% (IÎ 95%: 4,07, 15,83), p = 0,0011. Timpul median până la obţinerea celui mai bun răspuns şi durata mediană a celui mai bun răspuns la pacienţii care au atins RC sau RCp au fost de 4,3 luni şi respectiv 8,3 luni. Supravieţuirea fără progresia bolii a fost semnificativ mai mare pentru subiecţii din braţul decitabină, 3,7 luni (IÎ 95%: 2,7; 4,6), comparativ cu subiecţii din grupul OT, 2,1 luni (IÎ 95%: 1,9; 3.1), raportul de risc 0,75 (IÎ 95%: 0,62, 0,91), p = 0,0031. Aceste rezultate, precum şi alte obiective finale sunt prezentate în Tabelul 2.
Tabelul 2: Alte criterii finale de evaluare a eficacităţii pentru studiul DACO-016 (populaţia în ITT).
| Rezultate | Decitabină n = 242 | TC (grup combinat) n = 243 | Valoarea p |
|---|---|---|---|
| RC + RCp | 43 (17,8%) | 19 (7,8%) | 0,0011 |
| OR = 2.5 (1,40; 4,78)b | |||
| RC | 38 (15,7%) | 18 (7,4%) | - |
| SFEa | 3,5 (2,5; 4,1)b | 2,1 (1,9; 2,8)b | 0,0025 |
| HR = 0.75 (0,62; 0,90)b | |||
| SFPa | 3.7 (2,7; 4,6)b | 2.1 (1,9; 3,1)b | 0,0031 |
| HR = 0.75 (0,62; 0,91)b | |||
RC = remisiune completă; RCp = remisiune completă cu recuperare incompletă a trombocitelor, SFE = supravieţuire fără evenimente, SFP = supravieţuire fără progresia bolii, OR = odds ratio (raportul cotelor), RR = raport de risc- = Ne-evaluabil
a Raportată ca mediana lunilor
b Intervale de încredere 95%
Supravieţuirea globală şi ratele de remisiune completă în subgrupurile prespecificate, corelate cu boala (de exemplu, riscul citogenetic, scorul ECOG, vârsta, tipul de LMA şi valorile iniţiale ale numărului de celule blastice medulare) au fost în concordanţă cu rezultatele pentru populaţia generală în studiu.
Utilizarea decitabină ca terapie iniţială a fost, de asemenea, evaluată în cadrul unui studiu de fază II, deschis, cu un singur braţ de tratament (DACO-017) la 55 subiecţi cu vârsta > 60 ani, cu LMA conform clasificării OMS. Criteriul final principal de evaluare a fost rata de remisiune completă (RC), care a fost evaluată de o comisie independentă de experţi. Criteriul secundar de evaluare al studiului a fost supravieţuirea globală. decitabină a fost administrat ca perfuzie intravenoasă în decurs de 1 oră, în doză de 20 mg/m2 o dată pe zi, timp de 5 zile consecutiv, repetat la fiecare 4 săptămâni. În analiza ITT s-a observat o rată de 23,6% a RC (IÎ 95%: 13,2, 37%) la 13/55 din subiecţii trataţi cu decitabină. Timpul median până la atingerea RC a fost de 4,1 luni şi durata mediană a RC a fost de 18,2 luni. Mediana supravieţuirii globale, la populaţia ITT a fost de 7,6 luni (IÎ 95%: 5,7, 11,5).
Nu au fost evaluate eficacitatea şi siguranţa decitabină la pacienţii cu leucemie acută promielocitară sau leucemie a SNC.
Copii şi adolescenţi
Un studiu de fază I/II, deschis, multicentric a evaluat siguranţa şi eficacitatea decitabină în administrare secvenţială cu citarabină la copiii cu vârsta de 1 lună până la < 18 ani cu LMA recidivantă sau refractară la tratament. În acest studiu au fost înrolaţi în total 17 subiecţi, cărora li s-a administrat decitabină 20 mg/m2, din care 9 subiecţi au primit citarabină 1 g/m2 şi 8 subiecţi citarabină administrată în doză maximă tolerată de 2 g/m2. Toţi subiecţii au întrerupt tratamentul cu medicamentul în studiu. Motivele pentru întreruperea tratamentului au inclus progresia bolii (12 [70,6%] subiecţi), subiecți care au primit transplant (3 [17,6%]), decizia investigatorului (1 [5,9%]) şi „altele” (1 [5,9%]). Reacțiile adverse raportate au corespuns profilului de siguranţă cunoscut al decitabină la pacienţii adulţi (a se vedea pct. 4.8). Pe baza acestor rezultate negative, decitabină nu trebuie utilizat la copiii și adolescenții cu LMA cu vârsta < 18 ani deoarece nu a fost stabilită eficacitatea (a se vedea pct. 4.2).
5.2 Proprietăţi farmacocinetice
Parametrii de farmacocinetică (FC) populaţională pentru decitabină au fost cumulați din 3 studii clinice la 45 de pacienţi cu LMA sau sindrom mielodisplazic (SMD), utilizând schema terapeutică de 5 zile. În fiecare studiu, PC decitabinei a fost evaluată în a cincea zi a primului ciclu de tratament.
Distribuţie
Farmacocinetica decitabinei în urma administrării sub formă de perfuzie intravenoasă cu durata de 1 oră a fost descrisă ca având un model liniar bi-compartimental, caracterizată prin eliminarea rapidă din compartimentul central şi prin distribuţia relativ lentă din compartimentul periferic. Pentru un pacient tipic (greutate 70 kg/suprafaţa corporală de 1,73 m2), parametrii farmacocinetici ai decitabinei sunt enumeraţi în Tabelul 3 de mai jos.
Tabelul 3: Rezumatul analizei de farmacocinetică populaţională la un pacient obișnuit la care se administrează zilnic perfuzii de 1 oră cu decitabină în doză de 20 mg/m2 timp de 5 zile, o dată la fiecare 4 săptămâni
| Parametrua | Valoarea predictivă | IÎ 95% |
|---|---|---|
| Cmax (ng/ml) | 107 | 88,5 - 129 |
| AUCcum (ng.oră/ml) | 580 | 480 - 695 |
| t1/2 (min) | 68,2 | 54,2 – 79,6 |
| Vdss (l) | 116 | 84,1 - 153 |
| CL (l/oră) | 298 | 249 - 359 |
a Doza totală per ciclu de tratament a fost de 100 mg/m2
Decitabina demonstrează o FC liniară, iar în urma perfuziei intravenoase, concentraţiile la starea de echilibru sunt atinse în decurs de 0,5 ore. Pe baza modelului de simulare, parametrii farmacocinetici au fost independenţi de timp (adică nu s-au modificat de la un ciclu la altul) şi nu s-a observat acumulare în cazul acestei scheme de tratament. Legarea de proteinele plasmatice a decitabinei este neglijabilă (< 1%). Vdss al decitabinei la pacienţi cu cancer este mare, indicând distribuţia în ţesuturile periferice. Nu a existat nici o dovadă a influenţelor legate de vârstă, clearance-ul creatininei, bilirubina totală sau boală.
Metabolizare
La nivel intracelular, decitabina este activată prin fosforilare secvenţială prin intermediul activităţilor fosfokinazei la trifosfat aferent, care apoi este încorporat de către ADN polimeraza. Datele privind metabolizarea in vitro şi rezultatele studiului stării de echilibru a masei la om au indicat faptul că sistemul citocromului P450 nu este implicat în metabolizarea decitabinei. Calea principală de metabolizare este posibil dezaminarea prin citidin-dezaminaza din ficat, rinichi, epiteliu intestinal şi sânge. Rezultatele studiului stării de echilibru a masei la om au indicat că decitabina nemodificată în plasmă reprezintă aproximativ 2,4% din radioactivitatea plasmatică totală. Principalii metaboliţi circulanţi nu sunt consideraţi farmacologic activi. Prezenţa acestor metaboliţi în urină împreună cu clearance-ul corporal total înalt şi excreţia urinară scăzută pentru decitabina nemodificată în urină (~ 4% din doză) indică faptul că decitabina este metabolizată in vivo în mod considerabil. Studiile in vitro arată că decitabina nici nu inhibă şi nici nu induce enzimele citocromului CYP 450 în doze de până la 20 de ori concentraţia terapeutică plasmatică maximă (Cmax) observată. Prin urmare, nu sunt anticipate interacţiuni între medicamentele metabolizate de CYP, şi decitabina este puţin probabil să interacţioneze cu medicamente metabolizate pe aceste căi. În completare datele in vitro arată că decitabina este un substrat slab al gp-P.
Eliminare
Clearance-ul plasmatic mediu după administrarea intravenoasă la pacienţii cu cancer a fost > 200 l/ oră cu variabilitate moderată între subiecți (coeficientul de variaţie [CV] este aproximativ 50%). Excreţia de medicament nemodificat pare a avea un rol minor în eliminarea decitabinei.
Rezultatele unui studiu la starea de echilibru a masei cu 14C-decitabină marcată radioactiv la pacienţii cu cancer au arătat că 90% din doza administrată de decitabină (4% medicament nemodificat) se excretă în urină.
Informaţii suplimentare privind grupele speciale de pacienți
Efectele insuficienţei renale sau hepatice, sexului, vârstei sau rasei asupra farmacocineticii decitabinei nu au fost studiate în mod formal. Informaţiile asupra grupelor speciale de pacienți au fost derivate din date de farmacocinetică din cele 3 studii menţionate mai sus, şi dintr-un studiu de fază I la subiecţi cu SMD (N = 14; 15 mg/m2 x 3 ore, la fiecare 8 de ore x 3 zile).
Vârstnici
Analiza de PC pentru decitabină a arătat că după FC decitabinei nu este dependentă de vârstă (intervalul studiat între 40 până la 87 de ani; mediana 70 de ani).
Copii şi adolescenţi
Analiza de FP pentru decitabină a arătat că după luarea în considerare a greutăţii corporale, nu există nicio diferenţă de FC pentru decitabină la pacienți copii şi adolescenţi cu LMA faţă de pacienţii adulţi cu LMA sau SMD.
Sex
Analiza de FC pentru decitabină nu a arătat nici o diferenţă relevantă clinic între bărbaţi şi femei.
Rasă
Majoritatea pacienţilor studiaţi au fost de rasă caucaziană. Cu toate acestea, analiza farmacocinetică populaţională a decitabinei a indicat faptul că rasa nu a avut niciun efect evident asupra expunerii la decitabină.
Insuficienţă hepatică
FC pentru decitabină nu a fost studiată formal la pacienţii cu insuficienţă hepatică. Rezultatele unui studiu la starea de echilibru a masei la om şi experimentele in vitro menţionate mai sus au indicat faptul că este puţin probabil ca enzimele CYP să fie implicate în metabolizarea decitabinei. În completare, datele limitate privind analiza de FC populaţională nu indică dependenţa semnificativă a parametrului FC de concentraţia bilirubinei totale, chiar în prezența unor intervale largi ale nivelurilor de bilirubină totală. Prin urmare, este puţin probabil ca expunerea la decitabină să fie afectată la pacienţii cu insuficienţă hepatică.
Insuficienţă renală
PC decitabinei nu a fost studiată formal la pacienţii cu insuficienţă renală.
Analiza de FC populațională pe baza datelor limitate pentru decitabină nu indică dependenţe semnificative ale parametrilor FC în funcţie de clearance-ul creatininei normalizat, un indicator al funcţiei renale. Prin urmare, expunerea la decitabină nu puțin probabil să fie afectată la pacienţii cu insuficienţă renală.
5.3 Date preclinice de siguranţă
Nu s-au efectuat studii formale de carcinogenitate cu decitabină. Dovezile din literatura de specialitate indică faptul că decitabina are potenţial carcinogen. Datele disponibile din studiile in vitro şi in vivo oferă dovezi suficiente conform cărora decitabina are potenţial genotoxic. Datele din literatura de specialitate indică de asemenea faptul că decitabina are efecte adverse asupra tuturor aspectelor legate de ciclul de reproducere, care include fertilitatea, dezvoltarea embriofetală şi dezvoltarea postnatală. Studiile de toxicitate cu doze repetate pe mai multe cicluri la şobolan şi iepure au indicat faptul că toxicitatea primară a fost reprezentată de mielosupresie, care include efecte asupra măduvei osoase, care au fost reversibile la întreruperea tratamentului. S-a observat, de asemenea toxicitate gastrointestinală, și la bărbaţi atrofie testiculară care nu a fost reversibilă în timpul perioadelor de recuperare planificate. Administrarea decitabinei la puii/nou-născuţii de şobolan a demonstrat un profil de toxicitate generală comparabil cu cel de la şobolanii adulți. Nu a fost afectată dezvoltarea neurocomportamentală şi capacitatea de reproducere la puii/nou-născuţii de şobolan trataţi cu niveluri de doze care au indus mielosupresie. A se vedea pct. 4.2 pentru informaţii privind utilizarea la copii şi adolescenţi.
6. PROPRIETĂŢI FARMACEUTICE
6.1 Lista excipienţilor
Dihidrogenofosfat de potasiu (E340)
Hidroxid de sodiu (E524)
Acid clorhidric (pentru ajustarea pH-ului)
6.2 Incompatibilităţi
Acest medicament nu trebuie amestecat cu alte medicamente cu excepţia celor menţionate la pct. 6.6.
6.3 Perioada de valabilitate
Flacon nedeschis
30 luni
Soluţia reconstituită şi diluată
În interval de 15 minute de la reconstituire, concentratul (în 10 ml de apă sterilă pentru preparate injectabile) trebuie diluat suplimentar, utilizând lichide perfuzabile răcite în prealabil (2°C - 8°C). Această soluţie diluată preparată pentru perfuzie intravenoasă poate fi păstrată la o temperatură cuprinsă între 2°C și 8°C timp de până la maximum 3 ore, şi apoi timp de până la 1 oră la temperatura camerei (20°C – 25°C) înainte de administrare.
Din punct de vedere microbiologic, medicamentul trebuie utilizat în decursul perioadei recomandate mai sus. Este responsabilitatea utilizatorului să respecte durata şi condiţiile recomandate pentru păstrare şi să se asigure că reconstituirea s-a efectuat în condiţii de asepsie.
6.4 Precauţii speciale pentru păstrare
Acest medicament nu necesită condiții speciale de păstrare.
Pentru condițiile de păstrare ale medicamentului reconstituit și diluat, a se vedea secțiunea 6.3.
6.5 Natura şi conţinutul ambalajului
Flacon din sticlă de tip I, transparentă, incoloră, de 20 ml, prevăzut cu dop din cauciuc bromobutilic, sigilat cu capsă detașabilă din aluminiu și disc din polipropilenă.
Flacoanele pot fi sau nu acoperite cu un manșon de protecție (folie transparentă, incoloră, care înconjoară (tubular) flaconul pentru a oferi măsuri suplimentare de siguranță).
Mărimea ambalajului: 1 flacon.
6.6 Precauţii speciale pentru eliminarea reziduurilor şi alte instrucţiuni de manipulare
Recomandări pentru manipulare în condiţii de siguranţă
Trebuie evitat contactul pielii cu soluţia şi trebuie purtate mănuşi de protecţie. Trebuie aplicate procedurile standard pentru manipularea medicamentelor citotoxice.
Procedura de reconstituire
Pulberea trebuie reconstituită cu 10 ml de apă pentru preparate injectabile în condiţii de asepsie. După reconstituire, fiecare ml conţine aproximativ 5 mg de decitabină cu un pH între 6,7 şi 7,3. În interval de 15 minute de la reconstituire, soluţia trebuie diluată suplimentar cu soluții perfuzabile răcite (soluţie injectabilă de clorură de sodiu 9 mg/ml [0,9%] sau cu soluţie de glucoză injectabilă 5%) pentru a obţine o concentraţie finală între 0,15 şi 1,0 mg/ml. Pentru termenul de valabilitate şi precauţiile pentru păstrare după reconstituire, a se vedea pct. 6.3.
Decitabina nu trebuie administrat prin aceeaşi linie de perfuzie/ acces intravenos concomitent cu alte medicamente.
Eliminare
Acest medicament este numai pentru o singură utilizare. Orice medicament neutilizat sau material rezidual trebuie eliminat în conformitate cu reglementările locale.
7. DEŢINĂTORUL AUTORIZAŢIEI DE PUNERE PE PIAŢĂ
Teva B.V.
Swensweg 5
Haarlem, 2031 GA
Țările de Jos
8. NUMĂRUL(ELE) AUTORIZAŢIEI DE PUNERE PE PIAŢĂ
16565/2026/01
9. DATA PRIMEI AUTORIZĂRI SAU A REÎNNOIRII AUTORIZAŢIEI
Data primei autorizări: Aprilie 2026
10. DATA REVIZUIRII TEXTULUI
Aprilie 2026