MACITENTAN VIATRIS 10 mg
Rezumatul caracteristicilor produsului (RCP)
- 1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI
- 2. COMPOZIŢIA CALITATIVĂ ŞI CANTITATIVĂ
- 3. FORMA FARMACEUTICĂ
-
4. DATE CLINICE
- 4.1 Indicaţii terapeutice
- 4.2 Doze şi mod de administrare
- 4.3 Contraindicaţii
- 4.4 Atenţionări şi precauţii speciale pentru utilizare
- 4.5 Interacţiuni cu alte medicamente şi alte forme de interacţiune
- 4.6 Fertilitatea, sarcina şi alăptarea
- 4.7 Efecte asupra capacităţii de a conduce vehicule şi de a folosi utilaje
- 4.8 Reacţii adverse
- 4.9 Supradozaj
- 5. PROPRIETĂȚI FARMACOLOGICE
- 6. PROPRIETĂŢI FARMACEUTICE
- 7. DEŢINĂTORUL AUTORIZAŢIEI DE PUNERE PE PIAŢĂ
- 8. NUMĂRUL(ELE) AUTORIZAŢIEI DE PUNERE PE PIAŢĂ
- 9. DATA PRIMEI AUTORIZĂRI SAU A REÎNNOIRII AUTORIZAŢIEI
- 10. DATA REVIZUIRII TEXTULUI
1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI
Macitentan Viatris 10 mg comprimate filmate
2. COMPOZIŢIA CALITATIVĂ ŞI CANTITATIVĂ
Fiecare comprimat filmat conține macitentan 10 mg.
Excipienți cu efect cunoscut
Fiecare comprimat filmat conține manitol aproximativ 39 mg și lecitină din soia (E322) aproximativ 0,06 mg.
Pentru lista tuturor excipienţilor, vezi pct. 6.1.
3. FORMA FARMACEUTICĂ
Comprimat filmat.
Comprimat filmat, rotund, biconvex, de culoare albă până la aproape albă, cu diametrul de aproximativ 5,5 mm, marcat cu „10” pe o față și neted pe cealaltă față.
4. DATE CLINICE
4.1 Indicaţii terapeutice
Adulți
Macitentan Viatris, administrat în monoterapie sau în asociere, este indicat pentru tratamentul pe termen lung al hipertensiunii arteriale pulmonare (HTAP) la pacienţi adulţi aflaţi în clasa funcţională II sau III OMS (Organizația Mondială a Sănătății) (vezi pct. 5.1).
Copii și adolescenți
Macitentan Viatris, administrat în monoterapie sau în asociere, este indicat pentru tratamentul pe termen lung al hipertensiunii arteriale pulmonare (HTAP) la copii și adolescenți cu vârsta mai mică de 18 ani și greutate corporală ≥40 kg aflaţi în clasa funcţională II sau III OMS (vezi pct. 5.1).
4.2 Doze şi mod de administrare
Tratamentul trebuie iniţiat şi monitorizat numai de către un medic cu experienţă în tratarea HTAP.
Doze
Adulți, copii și adolescenți cu vârsta mai mică de 18 ani și greutate corporală de cel puțin 40 kg Doza recomandată este de 10 mg o dată pe zi. Macitentan Viatris trebuie luat în fiecare zi la aproximativ aceeași oră.
Dacă pacientul omite o doză de Macitentan Viatris, trebuie să i se spună pacientului să administreze această doză cât mai curând posibil, şi apoi să ia doza următoare la ora programată în mod obişnuit. Pacientului trebuie să i se spună să nu administreze două doze în acelaşi timp, dacă a fost omisă o doză.
Comprimatele filmate de 10 mg sunt recomandate numai la copii și adolescenți cu greutate corporală de cel puțin 40 kg.
Pentru copii și adolescenți care cântăresc mai puțin de 40 kg sunt disponibile comprimate dispersabile cu o concentrație mai mică, sub alte denumiri comerciale.
Grupe speciale de pacienți
Vârstnici
Nu este necesară ajustarea dozei la pacienţi cu vârsta peste 65 ani (vezi pct. 5.2).
Insuficienţă hepatică
Pe baza datelor farmacocinetice (PK), nu este necesară ajustarea dozei la pacienţi cu insuficienţă hepatică uşoară, moderată sau severă (vezi pct. 4.4 şi 5.2). Cu toate acestea, nu există experienţă clinică privind utilizarea de macitentan la pacienţii cu HTAP cu insuficienţă hepatică moderată sau severă. Tratamentul cu Macitentan Viatris nu trebuie iniţiat la pacienţi cu insuficienţă hepatică severă, sau cu valori ale aminotransferazelor hepatice semnificativ crescute clinic (de peste 3 ori limita superioară a valorilor normale (> 3 x LSVN); vezi pct. 4.3 şi 4.4).
Insuficienţă renală
Pe baza datelor farmacocinetice (PK), nu este necesară o ajustare a dozei la pacienţii cu insuficienţă renală. Nu există experienţă clinică privind utilizarea macitentan la pacienţii cu HTAP cu insuficienţă renală severă. Utilizarea Macitentan Viatris nu este recomandată la pacienţii care efectuează ședințe de dializă (vezi pct. 4.4 şi 5.2).
Copii și adolescenți
Nu au fost stabilite dozele și eficacitatea macitentan la copiii cu vârsta mai mică de 2 ani. Datele disponibile în prezent sunt prezentate la pct. 4.8, 5.1 și 5.2, dar nu se poate face nicio recomandare cu privire la doze.
Mod de administrare
Comprimatele filmate nu pot fi divizate și trebuie înghițite întregi, cu apă. Acest lucru se datorează filmului de acoperire al comprimatului, care protejează stomacul și permite medicamentului să acționeze corespunzător în organism. Divizarea comprimatului poate cauza iritarea stomacului sau scăderea eficacității medicamentului. Comprimatele pot fi administrate cu sau fără alimente.
4.3 Contraindicaţii
- Hipersensibilitate la substanţa activă, soia sau la oricare dintre excipienţii enumeraţi la pct. 6.1.
- Sarcina (vezi pct. 4.6).
- Femei cu potenţial fertil care nu folosesc metode contraceptive eficace (vezi pct. 4.4 şi 4.6).
- Alăptarea (vezi pct. 4.6).
- Pacienţi cu insuficienţă hepatică severă (cu sau fără ciroză hepatică) (vezi pct. 4.2).
- Valori iniţiale ale aminotransferazelor hepatice (aspartat aminotransferază (AST) şi/sau alaninaminotransferază (ALT) > 3 x limita superioară a valorilor normale, LSVN) (vezi pct. 4.2. şi 4.4).
4.4 Atenţionări şi precauţii speciale pentru utilizare
Raportul beneficiu/risc pentru macitentan nu a fost stabilit în cazul pacienţilor cu hipertensiune arterială pulmonară din clasa funcţională I OMS.
Funcţia hepatică
Creşterea aminotransferazelor hepatice (AST, ALT) a fost asociată cu HTAP şi cu antagoniştii receptorilor de endotelină (ARE). Tratamentul cu Macitentan Viatris nu trebuie iniţiat la pacienţii cu insuficienţă hepatică severă sau cu aminotransferaze crescute (> 3 × LSVN) (vezi pct. 4.2 şi 4.3) şi nu este recomandat la pacienţii cu insuficienţă hepatică moderată. Testele pentru enzimele hepatice trebuie efectuate înainte de iniţierea tratamentului cu Macitentan Viatris.
Pacienţii trebuie monitorizaţi îpentru semne de afectare hepatică şi se recomandă monitorizarea valorilor ALT şi AST în fiecare lună. Dacă apar creşteri persistente, inexplicabile, relevante clinic ale aminotransferazei, sau dacă aceste creşteri sunt însoţite de o creştere a bilirubinei > 2 × LSVN, sau de simptome clinice de afectare hepatică (de exemplu icter), tratamentul cu Macitentan Viatris trebuie întrerupt.
La pacienţii care nu au prezentat simptome clinice de afectare hepatică, reiniţierea tratamentului cu Macitentan Viatris poate fi luată în considerare după revenirea enzimelor hepatice la normal. Se recomandă controlul efectuat de către unmedic specialist hepatolog.
Concentraţia hemoglobinei
Scăderea concentrațiilor de hemoglobină a fost asociată cu o scădere a concentraţiei antagoniștilor receptorilor de endotelină (ARE) care includ macitentan (vezi pct. 4.8). În studiile controlate cu placebo, scăderea concentraţiei de hemoglobină asociate macitentan nu au fost progresive, s-au stabilizat după primele 4-12 săptămâni de tratament şi au rămas stabile în cursul tratamentului cronic. Au fost raportate cazuri de anemie care au necesitat transfuzia de masă sanguină asociată cu macitentan şi alți ARE. Nu este recomandată iniţierea tratamentului cu Macitentan Viatris la pacienţii cu anemie severă. Se recomandă măsurarea concentraţiilor hemoglobinei înainte de iniţierea tratamentului, iar testele să fie repetate în cursul tratamentului după cum este indicat clinic.
Boala veno-ocluzivă pulmonară
Au fost raportate cazuri de edem pulmonar în cazul utilizării vasodilatatoarelor (în principal prostacicline) la pacienţii cu boală veno-ocluzivă pulmonară. În consecinţă, dacă apar semne de edem pulmonar la pacienţii cu HTAP aflaţi sub tratament cu macitentan, trebuie luată în considerare posibilitatea prezenţei bolii veno-ocluzive pulmonare.
Utilizarea la femeile cu potenţial fertil
Tratamentul cu Macitentan Viatris trebuie iniţiat la femeile cu potenţial fertil numai după confirmarea absenței sarcinii, asigurarea consilierii adecvate privind contracepţia şi aplicarea metodelor contraceptive eficace (vezi pct. 4.3 şi pct. 4.6). Femeile nu trebuie să rămână gravide timp de 1 lună după întreruperea tratamentului cu Macitentan Viatris. Se recomandă efectuarea testului de sarcină în fiecare lună în timpul tratamentului cu Macitentan Viatris pentru a permite diagnosticul precoce de sarcină.
Administrarea concomitentă cu inductori puternici ai CYP3A4
În prezenţa inductorilor puternici ai CYP3A4 poate apărea reducerea eficacităţii macitentan. Trebuie evitată administrarea concomitentă de macitentan şi inductorii potenţi de CYP3A4 (de exemplu rifampicină, sunătoare, carbamazepină şi fenitoină) (vezi pct. 4.5).
Administrarea concomitentă cu inhibitori puternici ai CYP3A4
Este necesară prudență când macitentan este administrat concomitent cu inhibitori puternici ai CYP3A4 (de exemplu, itraconazol, ketoconazol, voriconazol, claritromicină, telitromicină, nefazodonă, ritonavir şi saquinavir) (vezi pct. 4.5).
Utilizarea concomitentă cu inhibitori moderaţi duali sau combinaţi de CYP3A4 şi de CYP2C9
Este necesară prudență când macitentan este administrat concomitent cu inhibitori duali moderaţi ai CYP3A4 şi CYP2C9 (de exemplu, fluconazol şi amiodaronă) (vezi pct. 4.5). De asemenea este necesară prudență când macitentan este administrat concomitent atât cu un inhibitor moderat al CYP3A4 (de exemplu, ciprofloxacină, ciclosporină, diltiazem, eritromicină, verapamil) cât şi cu un inhibitor moderat al CYP2C9 (de exemplu, miconazol, piperină) (vezi pct. 4.5).
Insuficienţă renală
Pacienţii cu insuficienţă renală pot prezenta un risc mai ridicat de hipotensiune arterială şi anemie în timpul tratamentului cu macitentan. Prin urmare, trebuie să fie luată în considerare monitorizarea tensiunii arteriale şi hemoglobinei. Nu există experienţă clinică privind utilizarea de macitentan la pacienţii HTAP cu insuficienţă renală severă. Se recomandă precauţie în cazul tratamentului la această populaţie de pacienţi. Nu există experienţă clinică privind utilizarea macitentan la pacienţii care fac dializă, prin urmare nu se recomandă administrarea Macitentan Viatris la această grupă de pacienţi (vezi pct. 4.2 şi 5.2).
Excipienţi cu efecte cunoscute
Macitentan Viatris conţine manitol care poate avea un efect uşor laxativ.
Macitentan Viatris conţine lecitină din soia. Dacă pacientul este hipersensibil la soia, Macitentan Viatris nu trebuie utilizat (vezi pct. 4.3).
Alţi excipienţi
Acest medicament conţine sodiu mai puţin de 1 mmol (23 mg) per comprimat filmat, adică practic „nu conţine sodiu”.
4.5 Interacţiuni cu alte medicamente şi alte forme de interacţiune
Studii in vitro
Citocromul P450 CYP3A4 este principala enzimă implicată în metabolizarea macitentan şi formarea metabolitului său activ, cu contribuţii minore ale enzimelor CYP2C8, CYP2C9 şi CYP2C19 (vezi pct. 5.2). Macitentan şi metabolitul său activ nu au efecte inhibitoare sau inductoare relevante clinic asupra enzimelor citocromului P450.
Macitentan şi metabolitul său activ nu sunt inhibitori ai transportorilor de recaptare hepatică sau renală la concentraţii relevante clinic, inclusiv a polipeptidelortransportoare de anioni organici (OATP1B1 şi OATP1B3). Macitentan şi metabolitul său activ nusunt substraturi relevante pentru OATP1B1 şi OATP1B3 dar pătrund în ficat prin difuziune pasivă.
Macitentan şi metabolitul său activ nu sunt inhibitori ai pompelor de eflux hepatic sau renal la concentraţii relevante clinic, inclusiv a proteinei de rezistenţă multiplă la medicamente (gp P , MDR- 1) şi transportatorii de extruziune multiplă a medicamentelor şi toxinelor (MATE1 şi MATE2-K). Macitentan nu este un substrat al gp P /MDR-1.
La concentraţii relevante clinic, macitentan şi metabolitul său activ nu interacţionează cu proteinele implicate în transportul sărurilor biliare hepatice, adică cu pompa de export asărurilor biliare (PESB) şi polipetida co-transportoare a taurocolatului, dependentă de sodiu (PTTN).
Studii in vivo
Inductori puternici ai CYP3A4
Tratamentul concomitent cu rifampicină 600 mg pe zi, un inductor puternic al CYP3A4, a redus expunerea la macitentan la starea de echilibru cu 79%, dar nu a afectat expunerea la metabolitul activ. Trebuie luată în considerare eficacitatea redusă a macitentan în prezenţa unui inductor puternic al CYP3A4, cum este rifampicina. Trebuie evitată administrarea concomitentă de macitentan şi inductorii puternici ai CYP3A4 (vezi pct. 4.4).
Ketoconazol
În prezenţa ketoconazolului 400 mg o dată pe zi, un inhibitor puternic al CYP3A4, expunerea la macitentan a crescut de aproximativ 2 ori. Creşterea estimată a fost de aproximativ 3 ori în prezenţa de ketoconazol200 mg de două ori pe zi, folosind modelarea farmacocinetică cu bază fiziologică (MFCF). Trebuie avute în vedere incertitudinile cu privire la această modelare . Expunerea la metabolitul activ al macitentan a fost redusă cu 26%. Este necesară prudență atunci când macitentan este administrat concomitent cu inhibitori puternici ai CYP3A4 (vezi pct. 4.4).
Fluconazol
În prezenţa fluconazolului 400 mg o dată pe zi, un inhibitor dual moderat al CYP3A4 şi CYP2C9, expunerea la macitentan poate creşte de aproximativ 3,8 ori, folosind MFCF. Cu toate acestea, nu a existat nicio modificare relevantă clinic a expunerii la metabolitul activ al macitentan. Trebuie avute în vedere incertitudinile cu privire la o astfel de MFCF. Este necesară prudență atunci când macitentan este administrat concomitent cu inhibitori duali moderaţi ai CYP3A4 şi CYP2C9 (de exemplu, fluconazol și amiodaronă) (vezi pct. 4.4).
De asemenea, este necesară prudență când macitentan este administrat concomitent atât cu un inhibitor moderat al CYP3A4 (de exemplu, ciprofloxacină, ciclosporină, diltiazem, eritromicină, verapamil) cât şi cu un inhibitor moderat al CYP2C9 (de exemplu, miconazol, piperină) (vezi pct. 4.4).
Warfarină
Macitentan administrat în doze multiple de 10 mg o dată pe zi nu a avut niciun efect asupra expunerii la S-warfarină (substrat al CYP2C9) sau R-warfarină (substrat al CYP3A4) după o doză unică de 25 mg warfarină. Efectul farmacodinamic al warfarinei asupra raportului internaţional normalizat (INR) nu a fost afectat de macitentan. Farmacocinetica macitentan şi a metabolitului său activ nu a fost afectată de warfarină.
Sildenafil
La starea de echilibru, expunerea la sildenafil în doză de 20 mg de trei ori pe zi a fost crescută cu 15% în cursul administrării concomitente de macitentan 10 mg o dată pe zi. Sildenafilul, un substrat al CYP3A4, nu a afectat farmacocinetica macitentan, existând în schimb o reducere cu 15% a expunerii la metabolitul activ al macitentan. Aceste modificări nu au fost considerate ca fiind relevante clinic. Într-un studiu controlat faţă de placebo la pacienţi cu HTAP, au fost demonstrate eficacitatea şi siguranţa tratamentului cu macitentan administrat concomitent cu sildenafil.
Ciclosporina A
Tratamentul concomitent cu ciclosporină A 100 mg de două ori pe zi, un inhibitor al CYP3A4 şi OATP, nu a afectat expunerea la macitentan şi metabolitul său activ, în starea de echilibru, într-o măsură relevantă clinic.
Contraceptive hormonale
Macitentan 10 mg o dată pe zi nu a afectat farmacocinetica unui contraceptiv oral (noretisteronă 1 mg şi etinilestradiol 35 μg).
Medicamente substrat al proteinei de rezistenţă la cancerul mamar (BCRP)
Administrarea de macitentan 10 mg o dată pe zi nu a afectat farmacocinetica unui medicament substrat al BCRP (riociguat 1 mg; rosuvastatină 10 mg).
Copii și adolescenți
Studiile privind interacțiunile au fost efectuate numai la adulți.
4.6 Fertilitatea, sarcina şi alăptarea
Utilizarea la femeile cu potenţial fertil/Contracepția la bărbați și femei
Tratamentul cu Macitentan Viatris trebuie iniţiat la femeile cu potenţial fertil numai după confirmarea absenței sarcinii, asigurarea consilierii adecvate privind contracepţia, şi utilizarea metodelor contraceptive eficace (vezi pct. 4.3 şi pct. 4.4). Femeile nu trebuie să rămână gravide timp de 1 lună după întreruperea tratamentului cu Macitentan Viatris. Se recomandă efectuarea testului de sarcină în fiecare lună în timpul tratamentului cu Macitentan Viatris pentru diagnosticul precoce al sarcinii.
Sarcina
Nu există date cu privire la utilizarea macitentan la femeile gravide. Studiile la animale au evidenţiat efecte toxice asupra funcţiei de reproducere (vezi pct. 5.3). Riscul potenţial la om este încă necunoscut. Macitentan Viatris este contraindicat în timpul sarcinii şi la femeile cu potenţial fertil care nu utilizează metode contraceptive eficace (vezi pct. 4.3).
Alăptarea
Nu se cunoaşte dacă macitentan se excretă în laptele uman. La şobolan, macitentan şi metaboliţii săi sunt excretaţi în lapte în perioada de lactaţie (vezi pct. 5.3). Riscul pentru copilul alăptat nu poate fi exclus. Administrarea Macitentan Viatris este contraindicată în timpul alăptării (vezi pct. 4.3).
Fertilitatea la bărbați
A fost observată prezența atrofiei tubilor seminiferi testiculari la animalele de sex masculin după tratamentul cu macitentan (vezi pct. 5.3). A fost observată scăderea numărului de spermatozoizi la pacienții cărora li s-au administrat ARE. Macitentan, ca și alți ARE, poate avea un efect advers asupra spermatogenezei la bărbați.
4.7 Efecte asupra capacităţii de a conduce vehicule şi de a folosi utilaje
Macitentan are influenţă minoră asupra capacităţii de a conduce vehicule și de a folosi utilaje. Nu s-au efectuat studii asupra capacității de a conduce vehicule și de a folosi utilaje. Cu toate acestea, pot apărea reacții adverse (de exemplu, cefalee, hipotensiune arterială) care pot afecta capacitatea de a conduce vehicule şi de a folosi utilaje (vezi pct. 4.8).
4.8 Reacţii adverse
Rezumatul profilului de siguranţă
Cele mai frecvent raportate reacţii adverse în studiul clinic SERAPHIN au fost rinofaringita (14%), cefaleea (13,6%) şi anemia (13,2%, vezi pct. 4.4).
Lista reacţiilor adverse sub formă tabelară
Siguranţa utilizării macitentan a fost evaluată în cadrul unui studiu de lungă durată, controlat cu placebo, la 742 pacienţi adulți și adolescenți cu HTAP simptomatică (studiul SERAPHIN). Durata medie de tratament a fost de 103,9 săptămâni în grupul cu administrare de macitentan 10 mg şi de 85,3 săptămâni în grupul cu administrare de placebo. Reacţiile adverse asociate cu macitentan obținute din acest studiu clinic sunt prezentate mai jos în formă de tabel. Reacțiile adverse observate după punerea pe piață sunt de asemenea incluse.
Frecvenţele de apariţie sunt definite astfel: foarte frecvente (≥ 1/10), frecvente (≥ 1/100 şi < 1/10), mai puţin frecvente (≥ 1/1000 şi < 1/100), rare (≥ 1/10000 şi < 1/1000), foarte rare (< 1/10000), cu frecvență necunoscută (care nu poate fi estimată din datele disponibile).
| Clasificare pe aparate, sisteme şi organe | Frecvenţă | Reacţii adverse |
|---|---|---|
| Infecţii şi infestări | Foarte frecvente | Rinofaringită |
| Foarte frecvente | Bronşită | |
| Frecvente | Faringită | |
| Frecvente | Gripă | |
| Frecvente | Infecţie de tracturinar | |
| Tulburări hematologice şi limfatice | Foarte frecvente | Anemie, scăderea hemoglobinei5 |
| Frecvente | Leucopenie6 | |
| Frecvente | Trombocitopenie7 | |
| Tulburări ale sistemului imunitar | Mai puțin frecvente | Reacţii de hipersensibilitate (de exemplu, angioedem, prurit, erupţie cutanată tranzitorie)1 |
| Tulburări ale sistemului nervos | Foarte frecvente | Cefalee |
| Tulburări vasculare | Frecvente | Hipotensiune arterială2, hiperemie facială |
| Tulburări respiratorii, toracice şi mediastinale | Frecvente | Congestie nazală1 |
| Tulburări hepatobiliare | Frecvente | Creșteri ale concentrațiilor aminotransferazelor4 |
| Tulburări ale sistemului reproducător și sânului | Frecvente | Hemoragie uterină crescută8 |
| Tulburări generale şi la nivelul locului de administrare | Foarte frecvente | Edem, retenţie de lichide3 |
1 Date provenite din studii grupate, controlate cu placebo.
8 Includ hemoragie menstruală abundentă, hemoragie uterină anormală, hemoragie intermenstruală, hemoragie uterină/vaginală, polimenoree și menstruație neregulată. Frecvența este stabilită în funcție de expunerea în cazul femeilor.
Descrierea reacţiilor adverse selectate
2 Hipotensiunea arterială a fost asociată cu utilizarea ARE, inclusiv macitentan. În studiul SERAPHIN, un studiu dublu-orb, de lungă durată, la pacienţi cu HTAP, hipotensiunea arterială a fost raportată la 7,0% şi 4,4% dintre pacienţii trataţi cu macitentan 10 mg, respectiv cu placebo. Aceasta corespunde cu 3,5 evenimente/100 pacienţi-an pentru macitentan 10 mg şi 2,7 evenimente/100 pacienţi-an pentru placebo.
3 Edemul/retenţia de lichide a fost asociat(ă) cu utilizarea ARE, inclusiv macitentan. În studiul SERAPHIN, un studiu dublu-orb, de lungă durată, la pacienţi cu HTAP, incidenţa reacţiilor adverse de edem, în grupurile tratate cu macitentan 10 mg şi cu placebo, a fost de 21,9% şi, respectiv, de 20,5%. Într-un studiu dublu-orb la pacienţi adulți cu fibroză pulmonară idiopatică, incidenţa reacţiilor adverse de edem periferic, în grupurile tratate cu macitentan şi cu placebo a fost de 11,8% şi, respectiv, de 6,8%. În două studii clinice în regim dublu-orb la pacienţi adulți cu ulcere digitale asociate cu scleroza sistemică, incidenţa reacţiilor adverse de edem periferic a fost cuprinsă între 13,4% şi 16,1% la grupurile tratate cu macitentan 10 mg şi între 6,2% şi 4,5% la grupurile tratate cu placebo.
Modificări ale rezultatelor de laborator
4 Aminotransferazele hepatice
Incidenţa creşterilor aminotransferazei (ALT/AST) > 3 × LSVN a fost 3,4% pentru macitentan 10 mg şi 4,5% pentru placebo în studiul SERAPHIN, un studiu dublu-orb la pacienţi cu HTAP. Creşterile > 5 × LSVN au apărut la 2,5% dintre pacienţii care au primit macitentan 10 mg faţă de 2% la pacienţii cu placebo.
5 Hemoglobină
În studiul SERAPHIN, un studiu dublu-orb desfăşurat la pacienţi cu HTAP, macitentan 10 mg a fost asociat cu o scădere medie a hemoglobinei cu 1 g/dl comparativ cu placebo. O scădere faţă de momentul iniţial a concentraţiei hemoglobinei sub 10 g/dl a fost raportată la 8,7% dintre pacienţii tratați cu macitentan 10 mg şi la 3,4% dintre pacienţii tratați cu placebo.
6 Leucocite
În studiul SERAPHIN, un studiu dublu-orb desfăşurat la pacienţi cu HTAP, administrarea macitentan 10 mg a fost asociată cu o scădere a numărului mediu de leucocite faţă de momentul iniţial de 0,7 × 109 /l, comparativ cu nicio schimbare la pacienţii cu placebo.
7 Trombocite
În studiul SERAPHIN, un studiu dublu-orb desfăşurat la pacienţi cu HTAP, administrarea macitentan 10 mg a fost asociată cu o scădere a numărului mediu de trombocite de 17 × 109 /l, comparativ cu o scădere medie de 11 × 109 /l la pacienţii cu placebo.
Siguranța pe termen lung
Dintre cei 742 de pacienți care au participat la studiul pivot în regim dublu orb SERAPHIN, 550 de pacienți au fost incluși în studiul de prelungire pe termen lung în regim deschis (RD). (Grupul RD a inclus 182 de pacienți care au continuat tratamentul cu macitentan 10 mg și 368 de pacienți cărora li sa administrat placebo sau macitentan 3 mg și au trecut la macitentan 10 mg.)
Monitorizarea pe termen lung a acestor 550 de pacienți pentru o durată medie de expunere de 3,3 ani și o expunere maximă de 10,9 ani a indicat un profil de siguranță similar cu cel descris mai sus în timpul etapei în regim dublu orb a studiului SERAPHIN.
Copii şi adolescenţi (cu vârsta cuprinsă între ≥2 ani și mai puțin de 18 ani)
Siguranța administrării de macitentan a fost evaluată în studiul TOMORROW, un studiu de fază 3 la pacienți copii și adolescenți cu HTAP. Un număr total de 72 de pacienți cu vârsta cuprinsă între ≥2 ani și mai puțin de 18 ani au fost randomizați pentru a li se administra macitentan. Vârsta medie la înrolare a fost de 10,5 ani (interval 2,1 ani-17,9 ani). Durata mediană a tratamentului în cadrul studiului randomizat a fost de 168,4 săptămâni (interval 12,9 săptămâni-312,4 săptămâni) în brațul de tratament cu macitentan.
În general, profilul de siguranță la copiii și adolescenții din această grupă de vârstă a fost în concordanță cu cel observat la populația adultă. În completare la reacțiile adverse enumerate în tabelul de mai sus, au fost raportate următoarele reacții adverse pediatrice: infecție a tractului respirator superior (31,9%), rinită (8,3%) și gastroenterită (11,1%).
Copii şi adolescenţi (vârsta ≥1 lună și mai puțin de 2 ani)
Alți 11 pacienți, cu vârsta ≥1 lună și mai puțin de 2 ani, au fost înrolați pentru a li se administra macitentan fără randomizare, 9 pacienți din brațul cu administrare în regim deschis al studiului TOMORROW și 2 pacienți japonezi din cadrul studiului PAH3001. La momentul înrolării, intervalul de vârstă al pacienților din studiul TOMORROW a fost de la 1,2 ani până la 1,9 ani, iar durata mediană a tratamentului a fost de 37,1 săptămâni (interval 7,0-72,9 săptămâni). La momentul înrolării, vârsta celor 2 pacienți din studiul PAH3001 a fost de 21 de luni și, respectiv, 22 de luni.
În general, profilul de siguranță la copiii din această grupă de vârstă a fost în concordanță cu cel observat la populația adultă și la copiii și adolescenții cu vârsta ≥2 ani și mai puțin de 18 ani, cu toate acestea, datele disponibile privind siguranța clinică sunt foarte limitate pentru a putea formula o concluzie fermă privind siguranța la copii cu vârsta mai mică de 2 ani.
Siguranţa şi eficacitatea macitentan la copii cu vârsta mai mică de 2 ani nu au fost stabilite (vezi pct. 4.2).
Raportarea reacţiilor adverse suspectate
Raportarea reacţiilor adverse suspectate după autorizarea medicamentului este importantă. Acest lucru permite monitorizarea continuă a raportului beneficiu/risc al medicamentului. Profesioniştii din domeniul sănătăţii sunt rugaţi să raporteze orice reacţie adversă suspectată la
Agenţia Naţională a Medicamentului şi a Dispozitivelor Medicale din România
Str. Aviator Sănătescu nr. 48, sector 1
Bucureşti 011478- RO e-mail: adr@anm.ro
Website: www.anm.ro
4.9 Supradozaj
Macitentan a fost administrat în doză unică de cel mult 600 mg la subiecţi adulți sănătoşi. Au fost observate reacţii adverse sub formă de cefalee, greaţă şi vărsături. În caz de supradozaj trebuie luate măsuri de susţinere standard, după cum este necesar. Din cauza gradului mare de legare de proteine al macitentan, este puţin probabil ca dializa să fie eficientă.
5. PROPRIETĂȚI FARMACOLOGICE
5.1 Proprietăţi farmacodinamice
Grupa farmacoterapeutică: antihipertensive, antihipertensive pentru hipertensiune arterială pulmonară, codul ATC: C02KX04.
Mecanism de acţiune
Endotelina (ET)-1 şi receptorii săi (ETA şi ETB) mediază efecte diferite cum sunt vasoconstricţia, fibroza, proliferarea, hipertrofia şi inflamaţia. În condiţii patologice cum este HTAP, sistemul ET local este stimulat şi implicat în procesele de hipertrofie vasculară şi deteriorare a organelor.
Macitentan este un antagonist puternic al receptorilor de endotelină, activ pe cale orală atât asupra receptorilor ETA, cât şi a receptorilor ETB, şi de aproximativ 100 ori mai selectiv in vitro pentru ETA, comparativ cu ETB. Macitentan prezintă o afinitate înaltă şi un grad mare de ocupare a receptorilor ET din celulele musculare netede din artera pulmonară la om. Acesta previne activarea mediată de endotelină a sistemelor celui de-al doilea mesager, care provoacă vasoconstricţie şi proliferarea celulelor musculare netede.
Eficacitate şi siguranţă clinică
Eficacitatea la pacienţii cu hipertensiune arterială pulmonară
Un studiu multicentric, dublu-orb, controlat faţă de placebo, pe grupuri paralele de studiu, axat pe evenimente, de fază 3, pentru determinarea rezultatelor (AC-055-302/SERAPHIN) a fost efectuat la 742 pacienţi cu HTAP simptomatică, care au fost randomizaţi în trei grupe de tratament (placebo [N = 250], 3 mg [N = 250] sau 10 mg [N = 242] de macitentan o dată pe zi), pentru a evalua efectul pe termen lung asupra morbidităţii şi mortalităţii.
La momentul iniţial, majoritatea pacienţilor înrolaţi (64%) erau trataţi cu o doză stabilă dintr-un tratament specific pentru HTAP, adică inhibitori de fosfodiesterază pe cale orală (61%) şi/sau prostanoizi pe cale inhalatorie/orală (6%).
Criteriul primar final de evaluare a fost timpul până la prima apariţie a unui eveniment de morbiditate sau mortalitate, până la finalul tratamentului dublu-orb, definit ca deces, septostomie atrială, transplant pulmonar, iniţierea tratamentului intravenos (i.v.) sau subcutanat (s.c.) cu prostanoizi sau alt mod de agravare a HTAP. Alt mod de agravare a HTAP a fost definit ca prezenţa tuturor următoarelor trei componente: o scădere stabilă a distanţei la testul de mers de 6 minute (6MWD) sau de cel puţin 15% 11 faţă de momentul iniţial, o agravare a simptomelor HTAP (agravarea clasei funcţionale OMS sau a insuficienţei cardiace drepte) şi necesitatea unui tratament nou pentru HTAP. Toate evenimentele au fost confirmate de o comisie de control independentă, care a lucrat în regim orb faţă de alocarea tratamentului.
Toţi pacienţii au fost urmăriţi până la sfârşitul studiului (end-of-study, EOS) pentru semnele vitale. EOS a fost declarat momentul în care a fost atins numărul predefinit de evenimente aparţinând criteriului primar final de evaluare. În perioada dintre sfârşitul tratamentului (end-of-treatment, EOT) şi EOS, pacienţii au putut primi macitentan 10 mg în regim deschis sau un tratament pentru HTAP alternativ. Mediana duratei globale a tratamentului dublu-orb a fost de 115 săptămâni (cel mult 188 săptămâni în cazul macitentan).
Vârsta medie pentru întregul lot de pacienţi a fost de 46 ani (între 12 şi 85 ani, incluzând 20 pacienţi sub 18 ani, 706 pacienţi între 18 şi 74 ani şi 16 pacienţi cu vârsta de 75 ani sau mai mare), majoritatea subiecţilor fiind de rasă caucaziană (55%) şi sex feminin (77%). Aproximativ 52%, 46% şi 2% dintre pacienţi au fost în clasa funcţională OMS II, III şi, respectiv, IV.
HTAP idiopatică sau congenitală a reprezentat cea mai frecventă etiologie la populaţia inclusă în studiu (57%), urmată de HTAP indusă de tulburările ţesutului conjunctiv (31%), HTAP asociată cu boală cardiacă congenitală simplă corectată (8%) şi HTAP asociată cu alte etiologii (medicamente şi toxine [3%] şi HIV [1%]).
Rezultatele evaluării
Tratamentul cu macitentan 10 mg a determinat o reducere cu 45% a riscului (raport de risc [RR] 0,55; 97,5% IÎ: 0,39 până la 0,76; logrank p < 0,0001) pentru criteriul compozit final de evaluare a morbimortalităţii până la EOT, comparativ cu placebo [Figura 1 şi Tabelul 1]. Efectul tratamentului a apărut precoce şi a fost de durată.
Eficacitatea macitentan 10 mg pentru criteriul primar final de evaluare a fost constantă pentru toate subgrupele de vârstă, sex, origine etnică, regiune geografică, etiologie, pentru monoterapie ca şi pentru combinaţia cu un alt tratament pentru HTAP, precum şi pentru toate clasele funcţionale OMS (I/II şi III/IV).
Figura 1 Estimările Kaplan-Meier privind primul eveniment de morbiditate-mortalitate în SERAPHIN
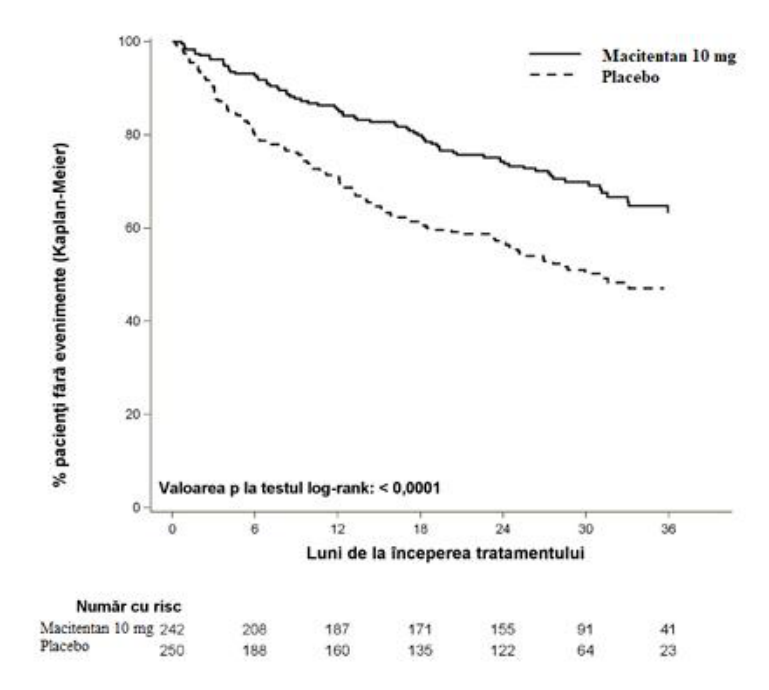
Tabelul 1: Rezumatul evenimentelor din categoria rezultatelor urmărite
| Criterii finale de evaluare şi statistici | Pacienţi cu evenimente | Compararea tratamentelor: Macitentan 10 mg comparativ cu Placebo | ||||
| Placebo (N=250) | Macitentan 10 mg (N=242) | Reducere a riscului absolut | Reducere a riscului relativ (97,5% IÎ) | RRa (97,5% IÎ) | Valoarea p logrank | |
| Eveniment de morbiditate b | 53% | 37% | 16% | 45% (24%; 61%) | 0,55 (0,39; 0,76) | < 0,0001 |
| Deces c n (%) | 19 (7,6%) | 14 (5,8%) | 2% | 36% (−42%; 71%) | 0,64 (0,29; 1,42) | 0,20 |
| Agravarea HTAP n (%) | 93 (37,2%) | 59 (24,4%) | 13% | 49% (27%; 65%) | 0,51 (0,35; 0,73) | < 0,0001 |
i.v./s.c. Iniţiere prostanoid n (%) | 6 (2,4%) | 1 (0,4%) | 2% | |||
a = bazat pe modelul Cox al riscului proporţional
b = % de pacienţi cu un eveniment la 36 luni = 100 × (1 - estimarea KM)
c = decesul de orice cauză până la EOT, indiferent de agravarea anterioară
Numărul deceselor de orice cauză până la EOS cu macitentan 10 mg a fost de 35, versus 44 cu placebo (RR 0,77; 97,5% IÎ: 0,46 până la 1,28).
Riscul de deces din cauza HTAP sau spitalizare pentru HTAP până la EOT a fost redus cu 50% (RR 0,50; 97,5% IÎ: 0,34 până la 0,75; logrank p < 0,0001) la pacienţii care au primit macitentan 10 mg (50 evenimente), comparativ cu placebo (84 evenimente). La 36 luni, 44,6% dintre pacienţii la care s-a administrat placebo şi 29,4% dintre pacienţii la care s-a administrat macitentan 10 mg (reducerea absolută a riscului = 15,2%) au fost spitalizaţi pentru HTAP sau au decedat dintr-o cauză legată de HTAP.
Criterii finale de evaluare a simptomelor
Capacitatea de efort fizic a fost evaluată ca un criteriu secundar final de evaluare. Tratamentul cu macitentan 10 mg a condus, în luna a 6-a, la o creştere medie, corectată faţă de placebo, a 6MWD de 22 metri (97,5% IÎ: 3 până la 41; p = 0,0078). Evaluarea 6MWD pe clase funcţionale a dus la o creştere medie, corectată faţă de placebo, de la momentul iniţial la luna a 6-a, de 37 metri (97,5% IÎ: 5 până la 69) la pacienţii din clasa funcţională III/IV şi de 12 metri (97,5% IÎ: -8 până la 33) la pacienţii din clasa funcţională I/II. Creşterea 6MWD obţinută cu macitentan a fost menţinută pe toată durata studiului.
Tratamentul cu macitentan 10 mg la luna a 6-a a crescut cu 74% șansa de îmbunătăţire a clasei funcţionale OMS comparativ cu placebo (raport de risc 1,74; 97,5% IÎ: 1,10 până la 2,74; p = 0,0063).
Macitentan 10 mg a îmbunătăţit calitatea vieţii conform evaluării efectuate prin chestionarul SF-36.
Criterii finale de evaluare hemodinamice
Parametrii hemodinamici au fost evaluaţi la un subset de pacienţi (placebo [N = 67], macitentan 10 mg [N = 57]) după 6 luni de tratament. Pacienţii trataţi cu macitentan 10 mg au obţinut o reducere mediană de 36,5% (97,5% IÎ: 21,7 până la 49,2%) a rezistenţei vasculare pulmonare şi o creştere de 0,58 l/min/m2 (97,5% IÎ: 0,28 până la 0,93 l/min/m2) a indicelui cardiac, comparativ cu placebo.
Date pe termen lung în HTAP
În cadrul monitorizării pe termen lung a unui număr de 242 de pacienți cărora li s-a administrat macitentan 10 mg în etapa dublu orb (DO) a studiului SERAPHIN, dintre care 182 au continuat cu macitentan în cadrul studiului de prelungire în regim deschis (RD) (RD SERAPHIN) (grup DO/RD), estimările Kaplan-Meier de supraviețuire la 1, 2, 5, 7 și 9 ani au fost de 95%, 89%, 73%, 63% și respectiv 53%. Durata mediană de monitorizare a fost de 5,9 ani.
Copii şi adolescenţi
Eficacitatea la copii și adolescenți se bazează în principal pe un exercițiu de extrapolare care corelează expunerea cu intervalul de doze eficiente la adulți, având în vedere asemănarea între boala la copii și boala adulți, precum și datele de suport privind eficacitatea și siguranța din studiul clinic de fază 3 TOMORROW descris mai jos.
Un studiu clinic de fază 3, multicentric, randomizat, în regim deschis, cu o perioadă de extensie în regim deschis cu un singur braț (TOMORROW) a fost efectuat pentru a evalua farmacocinetica, eficacitatea și siguranța administrării macitentan la copii și adolescenți cu HTAP simptomatică.
Criteriul primar final de evaluare a fost caracterizarea farmacocinetică (vezi pct. 5.2).
Principalul criteriu compozit secundar final de evaluare a fost timpul până la prima apariție a unui eveniment de progresie a bolii confirmat de Comitetul pentru evenimente clinice (CEC), care a avut loc între randomizare și vizita de la sfârșitul perioadei de bază (end of the core period, EOCP), definit ca deces (de orice cauză), septostomie atrială sau anastomoză Potts, înscriere pe lista de transplant pulmonar, spitalizare din cauza agravării HTAP sau agravarea clinicăa HTAP. Agravarea clinică a HTAP a fost definită ca: necesitatea sau inițierea unui nou tratament specific pentru HTAP sau a tratamentului intravenos cu diuretice sau administrarea continuă a oxigenului ȘI cel puțin una dintre următoarele: agravarea clasei funcţionale OMS sau apariția de novo sau agravarea sincopei sau apariția de novo sau agravarea a cel puțin 2 simptome ale HTAP sau apariția de novo sau agravarea semnelor de insuficiență cardiacă dreaptă care nu răspund la diuretice orale.
Alte criterii secundare finale de evaluare au inclus timpul până la prima spitalizare pentru HTAP confirmată de CEC, timpul până la decesul din cauza HTAP confirmat de CEC, ambele tipuri de evenimente survenite între randomizare și EOCP, timpul până la decesul de orice cauză între randomizare și EOCP, modificarea clasei funcționale OMS și datele privind nivelul fragmentului Nterminal al pro-peptidului natriuretic cerebral (NT-proBNP).
Copii şi adolescenţi (cu vârsta ≥2 ani și mai puțin de 18 ani)
Un număr total de 148 de pacienți cu vârsta cuprinsă între ≥2 ani și <18 ani au fost randomizați în raport 1:1 pentru a primi fie macitentan, fie tratament standard (Standard of Care, SoC). SoC a inclus tratament nespecific pentru HTAP și/sau tratament cu până la 2 medicamente specifice pentru HTAP (inclusiv un alt ERA) și a exclus tratamentul cu macitentan și prostanoizi i.v./s.c. Vârsta medie a fost de 9,8 ani (interval 2,1 ani-17,9 ani), cu 35 (23,6%) de pacienți cu vârsta ≥2 și <6 ani, 61 (41,2%) cu vârsta ≥6 și <12 ani și 52 (35,1%) cu vârsta ≥12 și <18 ani. Majoritatea pacienților au fost de rasă caucaziană (51,4%) și de sex feminin (59,5%). Pacienții au fost fie în clasa funcțională I OMS (25,0%), fie în clasa funcțională II OMS (56,1%), fie în clasa funcțională III OMS (18,9%).
HTAP idiopatică a fost cea mai frecventă etiologie în cadrul populației studiate (48,0%), urmată de HTAP asociată cu boală cardiacă congenitală corectată chirurgical (28,4%), HTAP cu boală cardiacă congenitală concomitentă (17,6%), HTAP congenitală (4,1%) și HTAP asociată cu tulburări ale ţesutului conjunctiv (2,0%). Boala cardiacă congenitală concomitentă a inclus, de regulă, doar defecte concomitente de mici dimensiuni, cum ar fi șuntul pre-tricuspidian, șuntul post-tricuspidian, defectul septal atrial, defectul septal ventricular, persistența de canal arterial, niciunul dintre acestea nefiind considerat cauza HTAP de orice grad.
Durata medie a tratamentului în cadrul studiului randomizat a fost de 183,4 săptămâni în brațul cu macitentan și de 130,6 săptămâni în brațul cu SoC.
Au fost observate mai puține evenimente legate de principalul criteriu secundar final de evaluare privind progresia bolii confirmată de CEC în brațul cu macitentan (21 evenimente/73 pacienți, 29%) în comparație cu brațul cu SoC (24 evenimente/75 pacienți, 32%), o reducere de 3% a riscului absolut. Raportul de risc a fost de 0,828 (IÎ 95% 0,460; 1,492; valoare p stratificată pe 2 fețe = 0,567). Tendința numerică spre beneficiu a fost determinată în principal de agravarea clinică a HTAP.
Alte analize secundare privind eficacitatea
Același număr de evenimente privind prima spitalizare pentru HTAP confirmată a fost observat în ambele grupuri (macitentan 11 vs. SoC 11; RR ajustat = 0,912, IÎ 95% = [0,393; 2,118]). În ceea ce privește intervalul de timp până la decesul din cauza HTAP confirmat de CEC și decesul de orice cauză, s-a observat un număr total de 7 decese (dintre care 6 din cauza HTAP confirmate de CEC) în grupul cu macitentan în comparație cu 6 decese (dintre care 4 din cauza HTAP confirmate de CEC) în grupul cu SoC.
A existat un procent numeric mai ridicat de pacienți aflați în clasa funcțională I sau II OMS în brațul cu macitentan, în comparație cu brațul cu SoC, raportat în Săptămâna 12 (88,7% în brațul cu macitentan, în comparație cu 81,7% în brațul cu SoC) și în Săptămâna 24 (90,0% în brațul cu macitentan, în comparație cu 82,5% în brațul cu SoC).
Tratamentul cu macitentan a avut tendința de a reduce nivelul inițial al NT-proBNP (pmol/l) în Săptămâna 12, în comparație cu brațul cu SoC (medie geometrică: 0,72; IÎ 95%: între 0,49 și 1,05), dar rezultatele nu au fost semnificative statistic (valoare p pe 2 fețe fiind 0,086). Tendința nesemnificativă clinic a fost mai puțin evidentă în Săptămâna 24 (medie geometrică: 0,97; IÎ 95%: între 0,66 și 1,43; valoare p pe 2 fețe fiind 0,884).
Rezultatele privind eficacitatea la pacienții cu vârstă între ≥ 2 ani și sub 18 ani au fost similare cu cele înregistrate la pacienții adulți.
Copii şi adolescenţi (vârsta ≥1 lună și mai puțin de 2 ani)
Un număr suplimentar de 11 pacienți, cu vârsta ≥1 lună și mai puțin de 2 ani, au fost înrolați pentru a primi macitentan, fără randomizare, 9 pacienți din brațul în regim deschis al studiului TOMORROW și 2 pacienți japonezi din studiul PAH3001. PAH3001 a fost un studiu de fază 3 multicentric, în regim deschis, cu un singur braț, cu copii și adolescenți japonezi (cu vârsta între ≥3 luni și <15 ani) cu HTAP, efectuat pentru a evalua farmacocinetica și eficacitatea administrării de macitentan.
La momentul inițial, 6 pacienți din studiul TOMORROW primeau tratament cu inhibitor de PDE5. La momentul înrolării, intervalul de vârstă al pacienților a variat între 1,2 ani-1,9 ani. Pacienții erau fie în clasa funcțională II OMS (4), fie în clasa funcțională I OMS (5). HTAP asociată unei boli cardiace congenitale a fost cea mai frecventă etiologie (5 pacienți), urmată de HTAP idiopatică (4 pacienți). Doza zilnică administrată inițial a fost de 2,5 mg macitentan, până când pacienții au împlinit vârsta de 2 ani. După o perioadă mediană de urmărire de 37,3 săptămâni, niciunul dintre pacienți nu a prezentat evenimente de progresie a bolii confirmate de CEC, spitalizări pentru HTAP confirmate de CEC, decese din cauza HTAP confirmate de CEC sau evenimente de deces de orice cauză. Nivelul NTproBNP a scăzut cu 42,9% (n=6) în Săptămâna 12, cu 53,2% (n=5) în Săptămâna 24 și cu 26,1% (n=6) în Săptămâna 36.
La momentul inițial, 1 pacient japonez din studiul PAH3001 primea tratament cu inhibitor de PDE5. Ambii pacienți japonezi erau de sex masculin și vârsta lor la momentul înrolării a fost de 21 și, respectiv, 22 de luni. Ambii pacienți erau în clasa funcțională Panama I și, respectiv, clasa funcțională Panama II, iar etiologia principală a fost HTAP survenită după corecția chirurgicală. În Săptămâna 24, s-a observat o reducere a nivelurilor inițiale ale NT-proBNP de -3,894 pmol/l și, respectiv, -16,402 pmol/l.
Corelarea expunerii cu pacienți adulți nu a fost stabilită la această grupă de vârstă (vezi pct. 4.2 și 5.2).
5.2 Proprietăţi farmacocinetice
Farmacocinetica macitentan şi a metabolitului său activ au fost studiate în principal la subiecţi adulți sănătoşi. Expunerea la macitentan la pacienţii cu HTAP a fost de aproximativ 1,2 ori mai mare decât la subiecţii sănătoşi. Expunerea la metabolitul activ, care este de aproximativ 5 ori mai puţin potent decât macitentan, a fost de aproximativ 1,3 ori mai mare la pacienţi decât la subiecţii sănătoşi.
Farmacocinetica macitentan la pacienţii cu HTAP nu a fost influenţată de severitatea bolii.
După administrare repetată, farmacocinetica macitentan este proporţională cu doza până la și inclusiv 30 mg.
Absorbție
Concentraţia plasmatică maximă de macitentan se obţine la aproximativ 8-9 ore de la administrarea de comprimate filmate și comprimate dispersabile. După aceea, concentraţia plasmatică de macitentan şi a metabolitului său activ scade lent, cu un timp aparent de înjumătăţire plasmatică prin eliminare de aproximativ 16 ore, respectiv 48 ore.
La subiecţii sănătoşi, expunerea la macitentan şi la metabolitul său activ nu este modificată de prezenţa alimentelor, prin urmare macitentan poate fi administrat cu sau fără alimente.
Distribuție
Macitentan şi metabolitul său activ sunt puternic legate de proteinele plasmatice (> 99%), în principal de albumină şi într-o mai mică măsură de alfa1-acid glicoproteină. Macitentan şi metabolitul său activ
ACT-132577 sunt bine distribuiţi în ţesuturi, după cum o indică volumul de distribuţie aparent (Vss/F) de aproximativ 50 l şi 40 l pentru macitentan, respectiv ACT-132577.
Metabolizare
Macitentan are patru căi primare de metabolizare. Depropilarea oxidativă a sulfamidei conduce la formarea unui metabolit activ farmacologic. Această reacţie depinde de sistemul citocromului P450, în principal CYP3A4 (aproximativ 99%), cu contribuţii minore ale CYP2C8, CYP2C9 şi CYP2C19. Metabolitul activ circulă în plasma umană şi poate contribui la efectul farmacologic. Alte căi metabolice generează produse care nu au activitate farmacologică. Pentru aceste căi, CYP2C9 joacă un rol predominant, cu contribuţii minore ale CYP2C8, CYP2C19 şi CYP3A4.
Eliminare
Macitentan este excretat numai după o metabolizare amplă. Calea de excreţie majoră este prin urină, resposabilă de aproximativ 50% din doză.
Grupe speciale de pacienţi
Nu există un efect relevant clinic al vârstei, sexului sau originii etnice asupra farmacocineticii macitentan şi metabolitului său activ.
Insuficienţa renală
Expunerea la macitentan şi la metabolitul său activ a fost crescută de 1,3, respectiv 1,6 ori la pacienţii adulți cu insuficienţă renală severă. Această creştere nu este considerată ca fiind relevantă clinic (vezi pct. 4.2 şi 4.4)
Insuficienţa hepatică
Expunerea la macitentan a scăzut cu 21%, 34% şi 6%, iar expunerea la metabolitul său activ cu 20%, 25% şi 25% la subiecţii adulți cu insuficienţă hepatică uşoară, moderată şi severă, respectiv. Această scădere nu este considerată ca fiind relevantă clinic (vezi pct. 4.2 şi 4.4).
Copii şi adolescenţi (cu vârsta cuprinsă între ≥1 lună și mai puțin de 18 ani)
Farmacocinetica macitentan și a metabolitului său activ, aprocitentan, a fost caracterizată la 47 de pacienți copii și adolescenți cu vârsta ≥2 ani și la 11 pacienți copii cu vârsta între ≥1 lună și mai puțin de 2 ani.
Regimurile de administare a dozelor de macitentan în funcție de greutatea corporală au dus la expuneri observate/simulate la pacienți copii și adolescenți cu vârsta de 2 ani și mai puțin de 18 ani, comparabile cu expunerile observate la pacienți adulți cu HTAP și la subiecți adulți sănătoși care au primit doza de 10 mg o dată pe zi.
În cazul copiilor din grupa de vârstă ≥1 lună și mai puțin de 2 ani nu s-au realizat expuneri la macitentan comparabile cu cele ale pacienților adulți cu HTAP care au primit doza de 10 mg o dată pe zi (vezi pct. 4.2)
5.3 Date preclinice de siguranţă
La câine, macitentan a determinat scăderea tensiunii arteriale la expuneri similare expunerii terapeutice la om. Îngroşarea intimei arterelor coronare a fost observată la o expunere de 17 ori mai mare decât expunerea la om, după un interval cuprins între 4 şi 39 săptămâni de tratament. Pe baza sensibilităţii specifice pentru fiecare specie şi a marjei de siguranţă, această constatare nu este considerată relevantă la om.
A fost observată creşterea greutăţii ficatului şi hipertrofia hepatocelulară la şoarece, şobolan şi câine după tratamentul cu macitentan. Aceste modificări au fost în mare măsură reversibile şi au fost considerate ca fiind adaptări fără reacții adverse ale ficatului la cererea metabolică crescută.
Macitentan a indus hiperplazie minimă sau uşoară în mucoasă şi infiltraţie inflamatorie în submucoasă la nivelul cavităţii nazale, în studiile de carcinogenitate la şoarece, la toate dozele. Nu a fost observată nicio modificare la nivelul cavităţii nazale în studiul de toxicitate la şoarece de 3 luni sau în studiile la şobolan şi câine.
Macitentan nu a fost genotoxic într-o baterie de teste standard in vivo și in vitro. Macitentan nu a fost fototoxic in vivo după o doză unică la expuneri de până la 24 de ori mai mari decât expunerea la om. Studiile de carcinogenitate cu durata de 2 ani nu au relevat un potenţial carcinogen la expuneri de 18 şi respectiv de 116 ori mai mari la şobolan şi şoarece față de expunerea la om.
Dilatarea tubilor seminiferi testiculari a fost observată în studii de toxicitate cronică la masculi de şobolan şi câine, cu margine de siguranţă de 11,6 și respectiv 5,8. Dilatarea tubulară a fost complet reversibilă. După 2 ani de tratament, atrofia tubilor testiculari a fost observată la şobolan la o expunere de 4 ori mai mare decât expunerea la om.
Hipospermatogeneza a fost observată într-un studiu de carcinogenitate pe durata vieții la şobolan şi în studii de toxicitate după doze repetate la câine la expuneri care au asigurat margine de siguranţă de 9,7 la şobolan şi 23 la câine. Marginea de siguranţă pentru fertilitate a fost 18 pentru masculii de şobolan şi 44 pentru femelele de șobolan. Nu au fost observate modificări testiculare la şoarece după tratament cu durata de până la 2 ani.
Macitentan a fost teratogen la iepure şi şobolan la toate dozele testate. La ambele specii au fost prezente malformații cardiovasculare şi de fuziune a arcului mandibular.
Administrarea de macitentan la femele de şobolan spre finalul gestației şi pe durata lactației, la niveluri de expunere maternă de 5 ori mai mare decât expunerea la om a scăzut rata de supravieţuire la pui şi a afectat capacitatea de reproducție a puilor care au fost expuși la macitentan la finalul perioadei de viaţă intrauterină şi prin lapte în perioada de lactație.
Tratamentul puilor de şobolan din ziua a 4-a până în ziua 114 postnatal a cauzat un declin al creşterii greutăţii corporale, ducând la efecte secundare asupra dezvoltării (încetinirea coborârii testiculare, scăderea reversibilă a lungimii oaselor lungi, estru prelungit). Au fost observate creşterea uşoară a pierderior pre- şi post-implantare, scăderea numărului mediu de pui, şi scăderea greutăţii testiculare şi epididimale la expuneri de 7 ori mai mari decât expunerea la om. Atrofia tubilor seminiferi testiculari, şi efecte minime asupra variabilelor reproducției şi morfologiei spermatozoizilor au fost înregistrate la expuneri de 3,8 ori mai mari decât expunerea la om.
6. PROPRIETĂŢI FARMACEUTICE
6.1 Lista excipienţilor
Nucleul comprimatului:
Manitol (E 421)
Celuloză microcristalină
Amidonglicolat de sodiu tip A
Povidonă K-30
Polisorbat 80
Stearat de magneziu
Film de acoperire:
Alcool (poli)vinilic
Dioxid de titan (E 171)
Lecitină (Soia) (E 322)
Talc
Gumă Xantan
6.2 Incompatibilităţi
Nu este cazul.
6.3 Perioada de valabilitate
3 ani
6.4 Precauţii speciale pentru păstrare
Acest medicament nu necesită condiții speciale de păstrare.
6.5 Natura şi conţinutul ambalajului
Blistere opace din PVC/PE/PVDC cu folie de acoperire din aluminiu.
Blister: 30 si 60 comprimate
Blister cu doze unitare: 30x1 comprimate
Este posibil ca nu toate mărimile de ambalaj să fie comercializate.
6.6 Precauţii speciale pentru eliminarea reziduurilor
Orice medicament neutilizat sau material rezidual trebuie eliminat în conformitate cu reglementările locale.
7. DEŢINĂTORUL AUTORIZAŢIEI DE PUNERE PE PIAŢĂ
Viatris Limited
Damastown Industrial Park
Mulhuddart, Dublin 15, DUBLIN, Irlanda
8. NUMĂRUL(ELE) AUTORIZAŢIEI DE PUNERE PE PIAŢĂ
16512/2026/01-02-03
9. DATA PRIMEI AUTORIZĂRI SAU A REÎNNOIRII AUTORIZAŢIEI
Data primei autorizări - Martie 2026
10. DATA REVIZUIRII TEXTULUI
Martie 2026