CALQUENCE 100 mg
Rezumatul caracteristicilor produsului (RCP)
- 1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI
- 2. COMPOZIŢIA CALITATIVĂ ŞI CANTITATIVĂ
- 3. FORMA FARMACEUTICĂ
-
4. DATE CLINICE
- 4.1 Indicaţii terapeutice
- 4.2 Doze şi mod de administrare
- 4.3 Contraindicaţii
- 4.4 Atenţionări şi precauţii speciale pentru utilizare
- 4.5 Interacţiuni cu alte medicamente şi alte forme de interacţiune
- 4.6 Fertilitatea, sarcina şi alăptarea
- 4.7 Efecte asupra capacităţii de a conduce vehicule şi de a folosi utilaje
- 4.8 Reacţii adverse
- 4.9 Supradozaj
- 5. PROPRIETĂŢI FARMACOLOGICE
- 80.
- 60.
- 40.
- 20.
- 0. 0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23
- 6. PROPRIETĂŢI FARMACEUTICE
- 7. DEŢINĂTORUL AUTORIZAŢIEI DE PUNERE PE PIAŢÃ
- 8. NUMĂRUL(ELE) AUTORIZAŢIEI DE PUNERE PE PIAŢĂ
- 9. DATA PRIMEI AUTORIZĂRI SAU A REÎNNOIRII AUTORIZAŢIEI
- 10. DATA REVIZUIRII TEXTULUI
- 1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI
- 2. COMPOZIŢIA CALITATIVĂ ŞI CANTITATIVĂ
- 3. FORMA FARMACEUTICĂ
-
4. DATE CLINICE
- 4.1 Indicaţii terapeutice
- 4.2 Doze şi mod de administrare
- 4.3 Contraindicaţii
- 4.4 Atenţionări şi precauţii speciale pentru utilizare
- 4.5 Interacţiuni cu alte medicamente şi alte forme de interacţiune
- 4.6 Fertilitatea, sarcina şi alăptarea
- 4.7 Efecte asupra capacităţii de a conduce vehicule şi de a folosi utilaje
- 4.8 Reacţii adverse
- 4.9 Supradozaj
- 5. PROPRIETĂŢI FARMACOLOGICE
- 80.
- 60.
- 40.
- 20.
- 0. 0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23
- 6. PROPRIETĂŢI FARMACEUTICE
- 7. DEŢINĂTORUL AUTORIZAŢIEI DE PUNERE PE PIAŢÃ
- 8. NUMĂRUL(ELE) AUTORIZAŢIEI DE PUNERE PE PIAŢĂ
- 9. DATA PRIMEI AUTORIZĂRI SAU A REÎNNOIRII AUTORIZAŢIEI
- 10. DATA REVIZUIRII TEXTULUI
1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI
Calquence 100 mg capsule
2. COMPOZIŢIA CALITATIVĂ ŞI CANTITATIVĂ
Fiecare capsulă conţine acalabrutinib 100 mg.
Pentru lista tuturor excipienţilor, vezi pct. 6.1.
3. FORMA FARMACEUTICĂ
Capsulă tare (capsulă).
Capsulă cu corp de culoare galbenă şi capac de culoare albastră, de mărimea 1 (20 mm), inscripţionată cu „ACA 100 mg” cu cerneală neagră.
4. DATE CLINICE
4.1 Indicaţii terapeutice
Calquence în monoterapie sau în asociere cu obinutuzumab este indicat pentru tratamentul pacienţilor adulţi cu leucemie limfocitară cronică (LLC) netratată anterior.
Calquence în asociere cu venetoclax cu sau fără obinutuzumab este indicat pentru tratamentul pacienților adulți cu leucemie limfocitară cronică (LLC) netratată anterior.
Calquence în monoterapie este indicat pentru tratamentul pacienţilor adulţi cu leucemie limfocitară cronică (LLC) cărora li s-a administrat cel puţin o terapie anterioară.
Calquence în asociere cu bendamustină și rituximab (BR) este indicat pentru tratamentul pacienților adulți cu limfom cu celule de manta (LCM) netratat anterior și care nu sunt eligibili pentru transplant autolog de celule stem (TACS).
Calquence în monoterapie este indicat pentru tratamentul pacienților adulți cu limfom cu celule de manta (LCM) recidivat sau refractar netratați anterior cu un inhibitor al tirozin kinazei Bruton (TKB).
4.2 Doze şi mod de administrare
Tratamentul cu acest medicament trebuie iniţiat şi supravegheat de un medic cu experienţă în utilizarea terapiilor antineoplazice.
Doze
Doza recomandată de Calquence în monoterapie sau în asociere cu alte medicamente este de 100 mg de acalabrutinib de două ori pe zi (echivalentul unei doze zilnice totale de 200 mg).
Intervalul de administrare a dozelor de Calquence este de aproximativ 12 ore.
În cazul administrării schemelor terapeutice în asociere, vă rugăm să consultați rezumatul caracteristicilor produsului al fiecărui medicament pentru informații referitoare la dozele recomandate (pentru detalii privind schemele terapeutice în asociere, vezi pct. 5.1).
Calquence în monoterapie sau în asociere cu obinutuzumab
Tratamentul cu Calquence în monoterapie sau în asociere cu obinutuzumab trebuie continuat până la progresia bolii sau până la apariţia toxicităţii inacceptabile.
Calquence în asociere cu venetoclax cu sau fără obinutuzumab
Tratamentul cu Calquence în asociere cu venetoclax cu sau fără obinutuzumab trebuie continuat până la progresia bolii, până la apariția toxicității inacceptabile sau până la finalizarea a 14 cicluri de tratament (fiecare ciclu are 28 de zile).
Calquence trebuie administrat în ziua 1 a primului ciclu, pentru un număr total de 14 cicluri de tratament. Venetoclax trebuie administrat în ziua 1 a ciclului 3, pentru un număr total de 12 cicluri, inițial în doză de 20 mg, care este crescută săptămânal la 50 mg, 100 mg, 200 mg și, la final, 400 mg.
Atunci când Calquence este administrat în asociere cu venetoclax și obinutuzumab, obinutuzumab trebuie administrat în doză de 100 mg în ziua 1 a ciclului 2, urmat de o doză de 900 mg care poate fi administrată în ziua 1 sau 2. Obinutuzumab trebuie administrat în doză de 1000 mg în zilele 8 și 15 ale ciclului 2, urmat de 1000 mg în ziua 1 începând cu ciclul 3 până la ciclul 7. Obinutuzumab se administrează pentru un număr total de 6 cicluri.
Calquence în asociere cu bendamustină și rituximab
Calquence trebuie administrat în ziua 1 a ciclului 1 de tratament (fiecare ciclu are 28 de zile) până la progresia bolii sau până la apariția toxicității inacceptabile. Bendamustina în doze de 90 mg/m2 se administrează în zilele 1 și 2 ale fiecărui ciclu, pentru un număr total de 6 cicluri de tratament. În ziua 1 a fiecărui ciclu se administrează rituximab în doze de 375 mg/m2, pentru un număr total de 6 cicluri de tratament. Pacienții care au obținut răspuns (răspuns parțial [RP] sau răspuns complet [RC]) după primele 6 cicluri de tratament pot să primească tratament de menținere cu rituximab în doze de 375 mg/m2 în ziua 1 a fiecărui al doilea ciclu și pentru maxim 12 doze suplimentare, începând cu ciclul 8 până la ciclul 30 de tratament.
Ajustarea dozelor
Reacţii adverse
Recomandările privind modificarea dozelor de Calquence în cazul reacţiilor adverse de grad ≥ 3 la pacienții cărora li se administrează Calquence în monoterapie, Calquence în asociere cu obinutuzumab sau Calquence în asociere cu venetoclax cu sau fără obinutuzumab sunt prezentate în tabelul 1.
Recomandările privind modificarea dozelor în cazul reacțiilor adverse de grad ≥ 3 la pacienții cărora li se administrează Calquence în asociere cu bendamustină și rituximab sunt prezentate în tabelul 2.
Tabelul 1. Ajustări recomandate ale dozelor în caz de reacţii adverse*
| Reacţie adversă | Apariţia reacţiei adverse | Modificarea dozei (doza de început = 100 mg la intervale aproximative de 12 ore) |
|---|---|---|
| Trombocitopenie de gradul 3 cu sângerare Trombocitopenie de gradul 4 Neutropenie de gradul 4 care persistă mai mult de 7 zile Toxicităţi non-hematologice de gradul 3 sau mai severe | Prima şi a doua | Se va întrerupe administrarea Calquence. Odată ce toxicitatea s-a remis la gradul 1 sau la valorile iniţiale, se poate relua administrarea Calquence în doze de 100 mg la intervale de aproximativ 12 ore. |
| A treia | Se va întrerupe administrarea Calquence. Odată ce toxicitatea s-a remis la gradul 1 sau la valorile iniţiale, se poate relua administrarea Calquence în doze de 100 mg o dată pe zi. | |
| A patra | Se va întrerupe definitiv tratamentul cu Calquence. |
*Reacţiile adverse au fost clasificate pe grade de severitate conform Criteriilor de terminologie comună pentru evenimentele adverse ale Institutului Naţional Oncologic (National Cancer Institute Common Terminology Criteria for Adverse Events, NCI CTCAE), versiunea 5.0.
Tabelul 2. Recomandări privind ajustarea dozelor în cazul reacțiilor adverse de grad ≥ 3* la pacienții tratați cu Calquence în asociere cu bendamustină și rituximab
| Reacție adversă | Modificarea dozei de bendamustinㆠ| Modificarea dozei de Calquence |
|---|---|---|
| Neutropenie | În cazul neutropeniei de grad 3 sau 4‡: se va întrerupe administrarea bendamustinei. Odată ce toxicitatea s-a remis la grad ≤ 2 sau la valorile inițiale, se poate relua administrarea bendamustinei în doze de 70 mg/m2. Se va întrerupe definitiv administrarea bendamustinei dacă sunt necesare reduceri suplimentare ale dozelor. | Se va întrerupe administrarea Calquence dacă neutropenia de grad 4 durează mai mult de 7 zile. Odată ce toxicitatea s-a remis la grad ≤ 2 sau la valorile inițiale, se poate relua administrarea Calquence cu doza de inițiere (la prima apariție a reacției adverse) sau cu frecvență redusă în doză de 100 mg zilnic, (la a doua sau a treia apariție a reacției adverse).¶ Se va întrerupe definitiv administrarea Calquence la a patra apariție a reacției adverse. |
| Trombocitopenie | În cazul trombocitopeniei de grad 3 sau 4: se va întrerupe administrarea bendamustinei. Odată ce toxicitatea s-a remis la grad ≤ 2 sau la valorile inițiale, se poate relua administrarea | Se va întrerupe administrarea Calquence în cazul apariției trombocitopeniei de grad 3 cu sângerare semnificativă sau trombocitopeniei de grad 4. Odată ce toxicitatea s-a remis la grad ≤ 2 sau la valorile inițiale, se poate relua administrarea Calquence cu doza de inițiere (la prima apariție a reacției adverse) sau cu frecvență redusă în doză de 100 mg zilnic |
| bendamustinei în doze de 70 mg/m2. Se va întrerupe definitiv administrarea bendamustinei dacă sunt necesare reduceri suplimentare ale dozelor. | (la a doua sau a treia apariție a reacției adverse).¶ În cazul apariției celei de-a treia reacții adverse de trombocitopenie cu sângerare semnificativă se va întrerupe definitiv administrarea Calquence. Se va întrerupe definitiv administrarea Calquence la a patra apariție a reacției adverse. | |
| Altă toxicitate hematologică de grad 4§ sau toxicitate de grad 3 care nu poate fi tratată | Se va întrerupe administrarea bendamustinei. Odată ce toxicitatea s-a remis la grad ≤ 2 sau la valorile inițiale, se va relua administrarea bendamustinei în doze de 70 mg/m2. Se va întrerupe definitiv administrarea bendamustinei dacă sunt necesare reduceri suplimentare ale dozelor. | Se va întrerupe administrarea Calquence. Odată ce toxicitatea s-a remis la grad ≤ 2 sau la valorile inițiale, se poate relua administrarea Calquence cu doza de inițiere (la prima apariție a reacției adverse) sau cu frecvență redusă în doză de 100 mg zilnic (la a doua sau a treia apariție a reacției adverse).¶ Se va întrerupe definitiv administrarea Calquence la a patra apariție a reacției adverse. |
| Toxicitate non-hematologică de grad 3 sau mai mare | Se va întrerupe administrarea bendamustinei. Odată ce toxicitatea s-a remis la grad 1 sau la valorile inițiale, se va relua administrarea bendamustinei în doze de 70 mg/m2. Se va întrerupe definitiv administrarea bendamustinei dacă sunt necesare reduceri suplimentare ale dozelor. | Se va întrerupe administrarea Calquence. Odată ce toxicitatea s-a remis la grad 2 sau la valorile inițiale, se va relua administrarea Calquence în doza de inițiere (la prima apariție a reacției adverse) sau cu frecvență redusă în doză de 100 mg zilnic (la a doua sau a treia apariție a reacției adverse).¶ Se va întrerupe definitiv administrarea Calquence la a treia apariție a reacției adverse. |
*Reacțiile adverse au fost clasificate pe grade de severitate conform Criteriilor de terminologie comună pentru evenimentele adverse ale Institutului Național Oncologic (National Cancer Institute Common Terminology Criteria for Adverse Events, NCI CTCAE) versiunea 4.03.
†În cazul în care nu au fost prezentate anumite toxicități în acest tabel, vă rugăm să consultați rezumatul caracteristicilor produsului bendamustină.
‡Se va lua în considerare administrarea factorilor de creștere mieloizi înainte de ajustarea dozelor de bendamustină. §Limfopenia de grad 4 este o reacție adversă așteptată în urma administrării tratamentului cu bendamustină și rituximab. Pot fi efectuate modificări ale dozelor doar dacă medicul investigator consideră aceste modificări ca fiind importante clinic, precum în cazul apariției infecțiilor recurente.
¶Dozele pot fi crescute la valorile inițiale conform deciziei medicului investigator dacă pacientul tolerează doze reduse pentru ≥ 4 săptămâni.
Pentru informații suplimentare privind tratamentul toxicităților vă rugăm să consultați rezumatul caracteristicilor produsului fiecărui medicament administrat în asociere cu Calquence.
Interacţiuni
În tabelul 3 sunt prezentate recomandările cu privire la utilizarea Calquence concomitent cu inhibitori sau inductori ai CYP3A şi cu medicamente care reduc secreţia acidului gastric (vezi pct. 4.5).
Tabelul 3. Utilizarea concomitent cu inhibitori sau inductori ai CYP3A şi cu medicamente care reduc secreţia acidului gastric
| Medicamentul administrat concomitent | Recomandări privind utilizarea Calquence | |
| Inhibitori CYP3A | Inhibitor puternic al CYP3A | Utilizarea concomitentă trebuie evitată. Dacă aceşti inhibitori se vor utiliza pe termen scurt (cum ar fi anti-infecţioasele, utilizate până la şapte zile), tratamentul cu Calquence trebuie întrerupt. |
Inhibitor moderat al CYP3A | Nu este necesară ajustarea dozelor. Pacienţii trebuie monitorizaţi cu atenţie pentru apariţia reacţiilor adverse în cazul administrării de inhibitori moderaţi ai CYP3A. | |
Inhibitor uşor al CYP3A | Nu este necesară ajustarea dozelor. | |
| Inductori ai CYP3A | Inductor puternic al CYP3A | Utilizarea concomitentă trebuie evitată. |
| Medicamente ce reduc secreţia de acid gastric | Inhibitori de pompă de protoni | Utilizarea concomitentă trebuie evitată. |
| Antagonişti ai receptorilor histaminergici H2 | Calquence trebuie administrat cu 2 ore înainte (sau la 10 ore după) administrarea unui antagonist al receptorilor histaminergici H2. | |
| Medicamente antiacide | Intervalul între administrările medicamentelor trebuie să fie de cel puţin 2 ore. |
Doze omise
Dacă pacientul omite să-și administreze o doză de Calquence şi au trecut mai mult de 3 ore de la momentul programat pentru administrare, pacientul trebuie instruit să ia următoarea doză conform schemei terapeutice obişnuite. Nu trebuie administrată o doză dublă de Calquence pentru a compensa doza omisă.
Grupe speciale de pacienți
Vârstnici
Nu este necesară ajustarea dozelor la pacienţi vârstnici (vârsta ≥ 65 ani) (vezi pct. 5.2).
Insuficienţă renală
Nu s-au efectuat studii clinice specifice la pacienţi cu insuficiență renală. Studiile clinice cu Calquence au inclus pacienţi cu insuficiență renală uşoară sau moderată. Nu este necesară ajustarea dozei la pacienţii cu insuficienţă renală uşoară sau moderată (clearance al creatininei mai mare de 30 ml/min). Trebuie menţinută o hidratare adecvată şi nivelurile serice ale creatininei trebuie monitorizate periodic. La pacienţii cu insuficiență renală severă (clearance al creatininei < 30ml/min) se va administra Calquence numai dacă beneficiile tratamentului depăşesc riscurile şi aceşti pacienţi trebuie monitorizați cu atenţie pentru apariția semnelor de toxicitate. Nu există date provenite de la pacienţii cu insuficienţă renală severă sau de la pacienţii dializaţi (vezi pct. 5.2).
Insuficiență hepatică
Nu există recomandări privind ajustarea dozelor la pacienţii cu insuficiență hepatică uşoară sau moderată (Clasificarea Child-Pugh clasa A, Clasificarea Child-Pugh clasa B sau valori ale bilirubinei totale de 1,5 până la 3 ori mai mari decât limita superioară a valorilor normale [LSVN] şi orice valori ale AST). Cu toate acestea, pacienţii cu insuficiență hepatică moderată trebuie atent monitorizaţi pentru semne de toxicitate. Nu se recomandă utilizarea Calquence la pacienţi cu insuficiență hepatică severă (Clasificarea Child-Pugh clasa C sau valori ale bilirubinei totale de >3 ori mai mari decât LSVN şi orice valori ale AST) (vezi pct. 5.2).
Boală cardiacă severă
Pacienţii cu boli cardiovasculare severe au fost excluşi din studiile clinice privind Calquence.
Copii și adolescenți
Siguranţa şi eficacitatea administrării Calquence la copii şi adolescenţi cu vârsta cuprinsă între 0 şi 18 ani nu au fost încă stabilite. Nu sunt disponibile date.
Mod de administrare
Calquence este indicat pentru administrare pe cale orală. Capsulele trebuie înghiţite întregi cu apă la aproximativ acelaşi moment în fiecare zi, împreună cu sau fără alimente (vezi pct. 4.5). Capsulele nu trebuie mestecate, dizolvate sau deschise, deoarece acest lucru poate modifica absorbţia medicamentului în organism.
4.3 Contraindicaţii
Hipersensibilitate la substanţa activă sau la oricare dintre excipienţii enumeraţi la punctul 6.1.
4.4 Atenţionări şi precauţii speciale pentru utilizare
Hemoragii
La pacienţii cu malignităţi hematologice trataţi cu Calquence în monoterapie sau în asociere cu alte medicamente au apărut evenimente hemoragice majore, inclusiv hemoragii gastro-intestinale sau la nivelul sistemului nervos central, unele dintre acestea având consecinţe letale. Aceste evenimente au survenit la pacienţi cu, dar şi fără trombocitopenie. Per ansamblu, evenimentele de sângerare au fost de tip mai puţin sever, cum ar fi apariţia echimozelor şi peteşiilor (vezi pct. 4.8).
Mecanismul de la baza evenimentelor de sângerare nu este bine înţeles.
Pacienţii trataţi cu agenţi antitrombotici pot fi expuşi unui risc crescut de apariţie a hemoragiilor. Dacă este necesar din punct de vedere medical, se recomandă prudență în administrare concomitent cu medicamente antitrombotice şi trebuie avută în vedere monitorizarea suplimentară a pacienţilor pentru apariția eventualelor semne de sângerare. Warfarina sau alți inhibitori ai vitaminei K nu trebuie administrați concomitent cu Calquence.
În cazul unei intervenţii chirurgicale, trebuie analizate beneficiile şi riscurile întreruperii tratamentului cu Calquence timp de cel puţin 3 zile înainte şi după intervenţie.
Infecții
La pacienţii cu malignităţi hematologice trataţi cu Calquence în monoterapie sau în asociere cu alte medicamente au apărut cazuri de infecţii grave (bacteriene, virale sau fungice), inclusiv evenimente letale. Aceste infecții au apărut preponderent în absența neutropeniei, infecția neutropenică fiind raportată la 10,1% dintre pacienții care au primit tratament în monoterapie și la 26,8% dintre pacienții care au primit tratamentul asociat. Au fost înregistrate cazuri de infecţii determinate de reactivarea virusului hepatitic B (VHB) şi a virusului herpes zoster (VHZ), cazuri de aspergiloză şi leucoencefalopatie multifocală progresivă (LMP) (vezi pct. 4.8).
Reactivare virală
S-au raportat cazuri de reactivare virală a hepatitei B la pacienţii cărora li se administra Calquence. Înainte de iniţierea tratamentului cu Calquence trebuie determinat statusul serologic al virusului hepatitic B (VHB). Dacă pacienţii sunt pozitivi la testul serologic pentru hepatita B, înainte de a începe tratamentul trebuie consultat un medic specialist în hepatologie şi pacientul trebuie monitorizat şi gestionat conform standardelor medicale locale pentru a se preveni reactivarea hepatitei B.
Au fost raportate cazuri de apariţie a leucoencefalopatiei multifocale progresive (LMP), inclusiv cazuri letale, după administrarea Calquence în contextul utilizării anterioare sau concomitente a unei terapii imunosupresoare. Medicii trebuie să ia în considerare LMP în diagnosticul diferenţial al pacienţilor cu semne sau simptome noi sau agravate de natură neurologică, cognitivă sau comportamentală. Dacă se suspicionează LMP, trebuie efectuate evaluările diagnostice corespunzătoare şi tratamentul cu Calquence trebuie întrerupt până la excluderea LMP. Dacă există orice suspiciune, se recomandă trimiterea pacientului la neurolog în vederea efectuării de teste diagnostice adecvate pentru LMP, inclusiv RMN, de preferinţă cu substanță de contrast, testarea lichidului cefalorahidian (LCR) pentru prezenţa ADN-ului viral al JC şi trebuie avută în vedere repetarea evaluărilor neurologice.
În cazul pacienţilor cu risc crescut de infecţii oportuniste trebuie avut în vedere tratamentul profilactic în conformitate cu standardul de îngrijire. Pacienţii trebuie monitorizaţi pentru apariţia semnelor şi simptomelor de infecţie şi trataţi după cum este adecvat din punct de vedere medical.
Citopenii
La pacienţii cu malignităţi hematologice trataţi cu Calquence în monoterapie sau în asociere cu alte medicamente au fost înregistrate citopenii de gradul 3 sau 4 , inclusiv neutropenie, anemie şi trombocitopenie apărute în cursul tratamentului. Valorile hemoleucogramei trebuie monitorizate după cum este indicat din punct de vedere medical (vezi pct. 4.8).
A doua malignitate primară
La pacienţii cu malignităţi hematologice trataţi cu Calquence în monoterapie sau în asociere cu alte medicamente a apărut şi a doua malignitate primară, inclusiv cancere cutanate sau non-cutanate. Cazurile de cancer cutanat au fost frecvent raportate. Pacienţii trebuie monitorizaţi pentru apariţia cancerelor cutanate şi sfătuiţi să se protejeze de expunerea la soare (vezi pct. 4.8).
Fibrilaţie atrială
În rândul pacienţilor cu malignităţi hematologice trataţi cu Calquence în monoterapie sau în asociere cu alte medicamente au apărut cazuri de fibrilaţie atrială/flutter atrial. Pacienţii trebuie monitorizaţi pentru apariţia simptomelor (de exemplu, palpitaţii, ameţeală, sincopă, durere toracică, dispnee) de fibrilaţie atrială şi flutter atrial, cu efectuarea unei ECG atunci când este indicat medical (vezi pct. 4.5 şi 4.2). La pacienţii care dezvoltă fibrilaţie atrială în timpul tratamentului cu Calquence, trebuie luată în considerare o evaluare detaliată a riscului de afecţiuni tromboembolice. La pacienţii cu risc înalt de afecţiuni tromboembolice, trebuie avut în vedere tratamentul strict controlat cu anticoagulante şi trebuie luate în considerare alte opţiuni terapeutice decât Calquence.
Sindrom de liză tumorală
Sindromul de liză tumorală (SLT) a fost raportat în cazul administrării Calquence. Pacienții la risc pentru SLT (de exemplu, prezența masei tumorale mari la momentul inițial) trebuie evaluați pentru un potențial risc de SLT și trebuie monitorizați atent conform indicațiilor clinice.
Boală pulmonară interstițială/pneumonită
Boala interstițială pulmonară (BIP)/pneumonita a fost raportată la pacienții cărora li s-a administrat tratament cu Calquence în asociere cu bendamustină și rituximab pentru LCM. Pacienții trebuie monitorizați pentru apariția simptomelor care indică prezența BIP/pneumonită (de exemplu, tuse, dispnee sau hipoxie) și în cazul manifestării BIP/pneumonitei pacienții trebuie tratați conform indicațiilor clinice.
Alte medicamente
Administrarea de inhibitori puternici ai CYP3A concomitent cu Calquence poate determina creşterea expunerii la acalabrutinib şi, în consecinţă, creşterea riscului de apariţie a toxicităţilor. Invers, administrarea de inductori ai CYP3A concomitent cu Calquence poate determina scăderea expunerii la acalabrutinib, cu riscul implicit de pierdere a eficacităţii. Administrarea concomitentă cu inhibitori puternici ai CYP3A trebuie evitată. Dacă acești inhibitori vor fi utilizați pe termen scurt (cum sunt medicamentele anti-infecțioase administrate până la 7 zile), tratamentul cu Calquence trebuie întrerupt. În cazul în care este administrat un inhibitor moderat al CYP3A, pacienţii trebuie monitorizaţi îndeaproape pentru apariţia semnelor de toxicitate (vezi pct. 4.2 şi 4.5). Utilizarea concomitentă cu inductori puternici ai CYP3A4 trebuie evitată datorită riscului de ineficacitate a tratamentului.
Calquence conţine sodiu
Acest medicament conţine sodiu mai puţin de 1 mmol (23 mg) per doză, adică practic „nu conţine sodiu”.
4.5 Interacţiuni cu alte medicamente şi alte forme de interacţiune
Acalabrutinib şi metabolitul său activ sunt metabolizaţi preponderent de enzima 3A4 a citocromului P450 (CYP3A4) şi ambii constituie substraturi pentru gp-P şi proteina de rezistență în cancerul mamar (breast cancer resistance protein, BCRP).
Substanţe active care pot creşte concentraţiile plasmatice de acalabrutinib
Inhibitori ai CYP3A/gp-P
Administrarea concomitentă cu un inhibitor puternic al CYP3A/gp-P (itraconazol în doză de 200 mg o dată pe zi, timp de 5 zile) a crescut valorile Cmax şi ASC pentru acalabrutinib de 3,9 ori şi, respectiv, de 5,0 ori la voluntari sănătoşi (N=17).
Administrarea concomitentă cu inhibitori puternici ai CYP3A/gp-P trebuie evitată. În cazul în care este necesară utilizarea pe termen scurt a inhibitorilor puternici de CYP3A/gp-P (de exemplu, ketaconazol, conivaptan, claritromicină, indinavir, itraconazol, ritonavir, telaprevir, posaconazol, voriconazol), tratamentul cu Calquence trebuie întrerupt (vezi pct. 4.2).
Administrarea concomitentă cu inhibitori moderați ai CYP3A (fluconazol 400 mg în doză unică sau isavuconazol 200 mg în doză repetată timp de 5 zile) la voluntari sănătoși a crescut valorile Cmax și ASC pentru acalabrutinib de 1,4 până la 2 ori, în timp ce valorile Cmax și ASC pentru metabolitul activ ACP- 5862 au scăzut de 0,65 ori până la 0,88 ori, comparativ cu administrarea acalabrutinib în monoterapie. Nu este necesară ajustarea dozei în cazul administrării concomitente cu inhibitori moderați ai CYP3A. Pacienții trebuie monitorizați îndeaproape pentru apariția reacțiilor adverse (vezi pct. 4.2).
Substanţe active care pot scădea concentraţiile plasmatice de acalabrutinib
Inductori ai CYP3A
Administrarea concomitentă cu un inductor puternic al CYP3A (rifampicină în doză de 600 mg o dată pe zi, timp de 9 zile) a scăzut valorile Cmax şi ASC pentru acalabrutinib cu 68% şi, respectiv, cu 77% la voluntari sănătoşi (N=24).
Administrarea concomitentă cu inductori potenţi ai activităţii CYP3A (de exemplu, fenitoină, rifampicină, carbamazepină) trebuie evitată. Utilizarea concomitentă a sunătorii trebuie evitată deoarece poate scădea în mod impredictibil concentraţiile plasmatice de acalabrutinib.
Medicamente care scad secreţia de acid gastric
Solubilitatea acalabrutinib scade odată cu creşterea pH-ului. Administrarea acalabrutinib concomitent cu un medicament antiacid (carbonat de calciu în doză de 1 g) a scăzut valoarea ASC a acalabrutinib cu 53% la subiecţi sănătoşi. Administrarea concomitent cu un inhibitor al pompei de protoni (omeprazol 40 mg timp de 5 zile) a scăzut ASC pentru acalabrutinib cu 43%.
Dacă este necesar tratamentul cu un medicament care scade aciditatea gastrică, se va lua în considerare un medicament antiacid (precum carbonatul de calciu) sau un antagonist al receptorilor histaminergici H2 (de exemplu, ranitidină sau famotidină). În cazul utilizării medicamentelor antiacide, intervalul între administrările medicamentelor trebuie să fie de cel puţin 2 ore (vezi pct. 4.2). În cazul utilizării de antagonişti ai receptorilor histaminergici H2, Calquence trebuie administrat cu 2 ore înainte de (sau la 10 ore după) antagonistul respectiv.
Din cauza efectului de lungă durată al inhibitorilor de pompă de protoni, este posibil ca decalajul în timp faţă de momentul administrării dozelor de inhibitori de pompă de protoni să nu elimine riscul de interacţiune cu Calquence și de aceea trebuie evitată administrarea concomitentă (vezi pct. 4.2).
Substanţe active ale căror concentraţii plasmatice pot fi modificate de Calquence
Substraturi pentru CYP3A
Pe baza datelor din studiile in vitro, nu se poate exclude posibilitatea ca acalabrutinib să inhibe CYP3A4 la nivel intestinal şi să crească astfel expunerea la medicamentele-substrat pentru CYP3A4 susceptibile a fi metabolizate de enzimele CYP3A din intestin. Se recomandă prudenţă în cazul utilizării acalabrutinib concomitent cu medicamente substrat pentru CYP3A4, cu interval terapeutic îngust şi care se administrează pe cale orală (precum ciclosporină, ergotamină, pimozidă).
Efectul acalabrutinib asupra medicamentelor substrat pentru CYP1A2
Studiile in vitro indică faptul că acalabrutinib induce activitatea CYP1A2. Administrarea acalabrutinib concomitent cu substraturi pentru CYP1A2 (de exemplu, teofilină, cafeină) poate scădea expunerea la aceste medicamente.
Efectele acalabrutinib şi ale metabolitului său activ, ACP-5862, asupra sistemelor transportoare de medicamente
Acalabrutinib poate creşte expunerea la medicamentele substrat pentru BCRP (de exemplu, metotrexatul) administrate simultan, prin inhibiţia BCRP de la nivel intestinal (vezi pct. 5.2). Pentru a reduce la minimum posibilitatea unei interacţiuni la nivel gastrointestinal (GI), medicamentele substrat pentru BCRP cu administrare pe cale orală şi cu interval terapeutic îngust, precum metotrexatul, trebuie administrate cu cel puţin 6 ore înainte sau după acalabrutinib.
ACP-5862 poate creşte expunerea la medicamentele substrat pentru MATE1 (de exemplu, metforminul) administrate simultan, prin inhibiţia MATE1 (vezi pct. 5.2). Pacienţii trataţi concomitent cu medicamente a căror eliminare depinde de transportorul MATE1 (precum metforminul) trebuie monitorizaţi pentru depistarea semnelor de modificare a tolerabilităţii ca urmare a creşterii expunerii la medicaţia administrată concomitent cu Calquence.
4.6 Fertilitatea, sarcina şi alăptarea
Femeile aflate la vârsta fertilă
Femeile cu potenţial fertil trebuie sfătuite să nu rămână însărcinate pe durata tratamentului cu Calquence.
Sarcina
Nu există date sau există un volum limitat de date privind utilizarea acalabrutinib la femeile gravide. Pe baza rezultatelor din studiile la animale, este posibil să existe un risc pentru făt asociat cu expunerea la acalabrutinib pe durata sarcinii. Au fost observate cazuri de distocie (travaliu dificil sau prelungit) la şobolan, iar administrarea la femele de iepure gestante s-a asociat cu reducerea creşterii fetale (vezi pct. 5.3).
Calquence nu trebuie utilizat pe durata sarcinii decât dacă starea clinică a femeii impune tratamentul cu acalabrutininb.
Alăptarea
Nu se cunoaşte dacă acalabrutinib se excretă în laptele uman. Nu există date cu privire la efectele acalabrutinib asupra sugarului sau asupra producţiei de lapte. Prezenţa acalabrutinib şi a metabolitului său activ a fost depistată în laptele matern al femelelor de şobolan. Nu se poate exclude un risc pentru sugari. Mamele aflate în perioada de alăptare sunt sfătuite să nu alăpteze pe durata tratamentului cu Calquence şi timp de încă 2 zile după administrarea ultimei doze.
Fertilitatea
Nu există date privind efectele Calquence asupra fertilităţii la om. În cadrul unui studiu non-clinic în care s-a administrat acalabrutinib la şobolani masculi şi femele nu au fost observate efecte adverse asupra parametrilor de fertilitate (vezi pct. 5.3).
4.7 Efecte asupra capacităţii de a conduce vehicule şi de a folosi utilaje
Calquence nu are nicio influenţă sau are influenţă neglijabilă asupra capacităţii de a conduce vehicule sau de a folosi utilaje. Cu toate acestea, pe durata tratamentului cu acalabrutinib au fost raportate stări de oboseală şi ameţeli, iar pacienţii care manifestă aceste simptome trebuie sfătuiţi să nu conducă vehicule sau să folosească utilaje decât după dispariţia acestora.
4.8 Reacţii adverse
Rezumatul profilului de siguranţă
Calquence în monoterapie
În rândul celor 1478 pacienţi trataţi cu Calquence în monoterapie, cele mai frecvente (≥ 20%) reacţii adverse induse de medicament (RAIM), de orice grad au fost infecţiile, diareea, cefaleea, durerea musculo-scheletică, echimozele, tusea, artralgia, fatigabilitatea, greața şi erupţiile cutanate tranzitorii. Cel mai frecvent raportate (≥ 5%) reacţii adverse induse de medicament cu severitate de grad ≥ 3 au fost infecţiile, leucopenia, neutropenia, anemia, a doua malignitate primară și trombocitopenia.
Calquence în asociere cu obinutuzumab
În rândul celor 223 de pacienţi trataţi cu Calquence în asociere cu obinutuzumab, cele mai frecvente (≥ 20%) RAIM de orice grad au fost infecţiile, durerea musculo-scheletică, diareea, cefaleea, leucopenia, neutropenia, tusea, fatigabilitatea, artralgia, greaţa, ameţeala şi constipația. Cel mai frecvent raportate (≥ 5%) reacţii adverse induse de medicament cu severitate de grad ≥ 3 au fost leucopenia, neutropenia, infecțiile, trombocitopenia şi anemia.
Calquence în asociere cu venetoclax
În rândul celor 291 de pacienți tratați cu Calquence în asociere cu venetoclax, cele mai frecvente (≥ 20%) RAIM de orice grad au fost infecțiile, neutropenia, cefaleea, echimozele, diareea și durerea musculoscheletică. Cel mai frecvent raportată (≥ 5%) RAIM cu severitate de grad ≥ 3 a fost neutropenia.
Calquence în asociere cu venetoclax și obinutuzumab
În rândul celor 284 de pacienți tratați cu Calquence în asociere cu venetoclax și obinutuzumab, cele mai frecvente (≥ 20%) RAIM de orice grad au fost infecțiile, neutropenia, cefaleea, echimozele, diareea, greața și durerea musculo-scheletică. Cel mai frecvent raportate (≥ 5%) RAIM cu severitate de grad ≥ 3 au fost neutropenia și trombocitopenia.
Calquence în asociere cu bendamustină și rituximab
În rândul celor 297 de pacienți tratați cu Calquence în asociere cu bendamustină și rituximab, cele mai frecvente (≥ 20%) RAIM de orice grad au fost neutropenia, greața, erupția cutanată tranzitorie, diareea, durerea musculo-scheletică, cefaleea, fatigabilitatea, vărsăturile, constipația, anemia și trombocitopenia. Cel mai frecvent raportate (≥ 5%) reacții adverse induse de medicament cu severitate de grad ≥3 au fost neutropenia, erupția cutanată tranzitorie, trombocitopenia, anemia, pneumonia, a doua malignitate primară, hipertensiunea arterială și a doua malignitate primară cu excepția cancerului cutanat de alt tip decât melanom.
Lista tabelară a reacțiilor adverse
Reacțiile adverse induse de medicament (RAIM) identificate în studiile clinice derulate la pacienți cărora li s-a administrat Calquence în monoterapie sau în terapie asociată pentru tratamentul malignităților hematologice sunt prezentate în tabelele următoare. Durata mediană a tratamentului cu Calquence în monoterapie conform datelor cumulate din studii a fost de 38,2 luni. Durata mediană a tratamentului cu Calquence în asociere cu bendamustină și rituximab a fost de 28,6 luni. Durata mediană a tratamentului cu Calquence la pacienții tratați cu Calquence în asociere cu venetoclax cu sau fără obinutuzumab a fost de 12,9 luni.
Reacţiile adverse induse de medicament sunt enumerate conform sistemului MedDRA de clasificare pe aparate, sisteme şi organe (ASO). În cadrul fiecărei clase de aparate, sisteme şi organe, reacţiile adverse sunt enumerate în funcţie de frecvenţă, începând cu cele mai frecvente. De asemenea, categoriile corespunzătoare de frecvenţă pentru fiecare RAIM sunt definite astfel: foarte frecvente (≥ 1/10); frecvente (≥ 1/100 şi < 1/10); mai puţin frecvente (≥ 1/1000 şi < 1/100); rare (≥ 1/10000 şi < 1/1000); foarte rare (< 1/10000); cu frecvenţă necunoscută (care nu poate fi estimată din datele disponibile). În cadrul fiecărei categorii de frecvenţă, reacţiile adverse sunt prezentate în ordinea descrescătoare a gravităţii.
Tabelul 4. Reacții adverse induse de medicament* la pacienţi cu malignităţi hematologice trataţi cu acalabrutinib în monoterapie (N=1478)
| ASO MedDRA | Termen MedDRA | Toate gradele (%) | Grad ≥ 3* (%) |
|---|---|---|---|
| Infecții și infestări | Infecții la nivelul tractului respirator superior | Foarte frecvente (25,8) | 1,2 |
| Pneumonie | Foarte frecvente (15,8) | 8,7 | |
| Sinuzită | Foarte frecvente (11,4) | 0,4 | |
| Infecţie la nivelul tractului urinar | Frecvente (9,9) | 1,8 | |
| Bronșită | Frecvente (9,7) | 0,6 | |
| Infecții cu virus herpetic† | Frecvente (9,1) | 0,9 | |
| Rinofaringită | Frecvente (8,3) | 0 | |
| Infecţii cu Aspergillus† | Mai puţin frecvente (0,7) | 0,6 | |
| Reactivarea hepatitei B | Mai puţin frecvente (0,4) | 0,3 | |
| Tumori benigne, maligne şi de tip nedeterminat | A doua malignitate primară (ADMP)† Cancer cutanat de alt tip decât melanomul† ADMP cu excepţia cancerului cutanat de alt tip decât melanomul† | Foarte frecvente (17,6) Frecvente (9,9) Frecvente (9,7) | 6,7 1,4 5,5 |
| Tulburări hematologice şi limfatice | Neutropenie† | Foarte frecvente (19,4) | 17,5 |
| Anemie† | Foarte frecvente (17,1) | 9,5 | |
| Trombocitopenie† | Foarte frecvente (11,5) | 6,2 | |
| Limfocitoză | Mai puţin frecvente (0,5) | 0,3 | |
| Tulburări metabolice și de nutriție | Sindrom de liză tumorală | Mai puţin frecvente (0,5) | 0,4 |
| Tulburări ale sistemului nervos | Cefalee | Foarte frecvente (36,5) | 1,2 |
| Amețeală | Foarte frecvente (13,9) | 0,1 | |
| Tulburări cardiace | Fibrilaţie atrială/flutter atrial† † | Frecvente (7,4) | 2,3 |
| Tulburări vasculare | Echimoze† Contuzii Peteşii Echimoze | Foarte frecvente (30,9) Foarte frecvente (20,7) Frecvente (8,9) Frecvente (5,7) | 0 0 0 0 |
| Hemoragie/hematom† Hemoragie gastro-intestinală Hemoragie intracraniană | Foarte frecvente (16,3) Mai puțin frecvente (0,9) Mai puțin frecvente (0,1) | 3,2 0,7 0,1 | |
| Hipertensiune arterialㆠ| Foarte frecvente (11,9) | 4,9 | |
| Epistaxis | Frecvente (8,0) | 0,3 | |
| Diaree | Foarte frecvente (36,7) | 2,6 | |
| Tulburări gastro- intestinale | Greaţă | Foarte frecvente (21,8) | 0,8 |
| Constipaţie | Foarte frecvente (15,2) | 0,1 | |
| Durere abdominalㆠ| Foarte frecvente (14,5) | 1,2 | |
| Vărsături | Foarte frecvente (14,0) | 0,7 | |
| Afecțiuni cutanate și ale țesutului subcutanat | Erupţie cutanată tranzitorie† | Foarte frecvente (20,3) | 0,9 |
| Tulburări musculo-scheletice şi ale ţesutului conjunctiv | Durere musculo-scheleticㆠ| Foarte frecvente (31,9) | 1,8 |
| Artralgie | Foarte frecvente (24,0) | 0,9 | |
| Tulburări generale şi la nivelul locului de administrare | Fatigabilitate | Foarte frecvente (23,6) | 2,0 |
| Astenie | Frecvente (7,0) | 0,9 | |
| Investigaţii diagnostice§ (constatări bazate pe rezultatele testelor) | Scăderea valorilor hemoglobinei± | Foarte frecvente (47,4) | 10,8 |
| Scăderea numărului absolut de neutrofile (NAN)± | Foarte frecvente (43,9) | 24,0 | |
| Scăderea numărului de trombocite± | Foarte frecvente (36,9) | 9,5 |
*Conform Criteriilor de terminologie comună pentru evenimentele adverse ale Institutului Național Oncologic (National Cancer Institute Common Terminology Criteria for Adverse Events, NCI CTCAE), versiunea 4.03. †Include mai mulți termeni pentru RAIM.
±Reprezintă incidența valorilor rezultatelor de laborator, nu a evenimentelor adverse raportate.
§Prezentate pe grade de severitate CTCAE.
Tabelul 5. Reacţii adverse induse de medicament* la pacienţii cu malignităţi hematologice trataţi cu acalabrutinib ca terapie asociată (N=1095)
| Calquence + obinutuzumab N=223 | Calquence + BR N=297 | Calquence + venetoclax N=291 | Calquence + venetoclax + obinutuzumab N=284 | |||||
| ASO MedDRA și Termen MedDRA | Toate gradele (%) | Grad ≥ 3* (%) | Toate gradele (%) | Grad ≥ 3* (%) | Toate gradele (%) | Grad ≥ 3* (%) | Toate gradele (%) | Grad ≥ 3* (%) |
| Infecții și infestări | ||||||||
|---|---|---|---|---|---|---|---|---|
| Infecții ale tractului respirator superior | Foarte frecvente (31,4) | 1,8 | Foarte frecvente (18,2) | 0,3 | Frecvente (8,2) | 0,3 | Frecvente (6,3) | 0 |
| Sinuzită | Foarte frecvente (15,2) | 0,4 | Frecvente (6,4) | 0 | Frecvente (2,7) | 0 | Frecvente (2,5) | 0 |
| Rinofaringită | Foarte frecvente (13,5) | 0,4 | Frecvente (5,4) | 0 | Frecvente (1,4) | 0 | Frecvente (1,1) | 0 |
| Infecții ale tractului urinar | Foarte frecvente (13) | 0,9 | Foarte frecvente (11,1) | 1,7 | Frecvente (3,1) | 0 | Frecvente (6,0) | 0,4 |
| Pneumonie | Foarte frecvente (10,8) | 5,4 | Foarte frecvente (16,2) | 8,8 | Frecvente (3,8) | 1,4 | Frecvente (5,3) | 3,9 |
| Bronșită | Frecvente (9,9) | 0 | Frecvente (6,4) | 0,3 | Frecvente (2,1) | 0 | Frecvente (2,5) | 0 |
| Infecții cu virus herpetic† | Frecvente (6,7) | 1,3 | Foarte frecvente (12,8) | 1,0 | Frecvente (4,8) | 0 | Frecvente (3,5) | 0,4 |
| Leucoencefalopatie multifocală progresivă | Mai puțin frecvente (0,4) | 0,4 | Cu frecvență necunoscută | 0 | Cu frecvență necunoscută | 0 | Cu frecvență necunoscută | 0 |
| Reactivarea hepatitei B | Mai puțin frecvente (0,9) | 0,1 | Frecvente (1,3) | 0,3 | Cu frecvență necunoscută | 0 | Cu frecvență necunoscută | 0 |
Infecții cu Aspergillus† | Cu frecvență necunoscută | 0 | Mai puțin frecvente (0,3) | 0,3 | Cu frecvență necunoscută | 0 | Mai puțin frecvente (0,4) | 0,4 |
| Tumori benigne, maligne și de tip nedeterminat | ||||||||
A doua malignitate primară†(ADMP) Cancer cutanat de alt tip decât melanom† ADMP cu excepția cancerului cutanat de alt tip decât melanom† | Foarte frecvente (13) Frecvente (7,6) Frecvente (6,3) | 4,0 0,4 3,6 | Foarte frecvente (17,8) Foarte frecvente (11,1) Frecvente (9,8) | 7,4 2,0 5,4 | Frecvente (5,2) Frecvente (3,1) Frecvente (2,7) | 1,7 0 1,7 | Frecvente (4,2) Frecvente (1,8) Frecvente (2,5) | 1,8 0,4 1,4 |
| Tulburări hematologice și limfatice | ||||||||
| Neutropenie† | Foarte frecvente (31,8) | 30 | Foarte frecvente (54,9) | 50,2 | Foarte frecvente (37,1) | 32,3 | Foarte frecvente (50,4) | 46,1 |
| Trombocitopenie† | Foarte frecvente (13,9) | 9 | Foarte frecvente (22,9) | 9,8 | Frecvente (5,8) | 2,1 | Foarte frecvente (12,3) | 9,2 |
| Anemie† | Foarte frecvente (11,7) | 5,8 | Foarte frecvente (24,2) | 9,4 | Frecvente (6,9) | 3,8 | Frecvente (4,6) | 2,1 |
| Limfocitoză | Mai puțin frecvente (0,4) | 0,4 | Mai puțin frecvente (0,7) | 0 | Cu frecvență necunoscută | 0 | Mai puțin frecvente (0,7) | 0,4 |
| Tulburări metabolice și de nutriție | ||||||||
| Sindrom de liză tumorală | Frecvente (1,8) | 1,3 | Frecvente (1,3) | 1,3 | Mai puțin frecvente (0,3) | 0,3 | Mai puțin frecvente (0,4) | 0,4 |
| Tulburări ale sistemului nervos | ||||||||
| Cefalee | Foarte frecvente (43) | 0,9 | Foarte frecvente (30,3) | 1,3 | Foarte frecvente (35,1) | 1,4 | Foarte frecvente (28,2) | 0,4 |
| Amețeală | Foarte frecvente (23,8) | 0 | Foarte frecvente (14,5) | 0,7 | Frecvente (5,5) | 0 | Frecvente (6,7) | 0 |
| Tulburări cardiace | ||||||||
| Fibrilație atrială/flutter atrial† | Frecvente (3,1) | 0,9 | Frecvente (6,7) | 4,0 | Mai puțin frecvente (0,7) | 0,3 | Frecvente (2,1) | 0,7 |
| Tulburări vasculare | ||||||||
Echimoze† Contuzii Peteșii Echimoze | Foarte frecvente (38,6) Foarte frecvente (27,4) Foarte frecvente (11,2) Frecvente (3,1) | 0 0 0 0 | Foarte frecvente (14,1) Foarte frecvente (11,1) Frecvente (2,0) Frecvente (3,0) | 0,3 0 0 0,3 | Foarte frecvente (20,6) Foarte frecvente (14,1) Frecvente (4,8) Frecvente (2,7) | 0 0 0 0 | Foarte frecvente (21,8) Foarte frecvente (16,2) Frecvente (5,3) Frecvente (3,9) | 0 0 0 0 |
Hemoragie/hematom Hemoragie gastro- intestinală Hemoragie intracraniană | Foarte frecvente (17,5) Frecvente (3,6) Mai puțin frecvente (0,9) | 1,3 0,9 0 | Foarte frecvente (15,5) Mai puțin frecvente (0,3) Cu frecvență necunoscută | 1,0 0 0 | Frecvente (8,9) Mai puțin frecvente (0,7) Cu frecvență necunoscută | 0,7 0,3 0 | Frecvente (8,5) Cu frecvență necunoscută Cu frecvență necunoscută | 1,1 0 0 |
| Hipertensiune arterialㆠ| Foarte frecvente (13,5) | 3,6 | Foarte frecvente (12,5) | 5,7 | Frecvente (4,1) | 2,7 | Frecvente (3,9) | 2,1 |
| Epistaxis | Frecvente (8,5) | 0 | Frecvente (2,7) | 0 | Frecvente (1,7) | 0 | Frecvente (4,2) | 0 |
| Tulburări respiratorii, toracice și mediastinale | ||||||||
| Pneumonită± | - | - | Frecvente (2,4) | 0,3 | - | - | - | - |
| Tulburări gastro-intestinale | ||||||||
| Diaree | Foarte frecvente (43,9) | 4.5 | Foarte frecvente (37,4) | 3,0 | Foarte frecvente (32,6) | 1,7 | Foarte frecvente (36,3) | 1,4 |
| Greață | Foarte frecvente (26,9) | 0 | Foarte frecvente (42,8) | 1,3 | Foarte frecvente (14,8) | 0 | Foarte frecvente (21,8) | 0,7 |
| Constipație | Foarte frecvente (20,2) | 0 | Foarte frecvente (24,6) | 1,0 | Frecvente (6,5) | 0,3 | Frecvente (8,1) | 0 |
| Vărsături | Foarte frecvente (19,3) | 0,9 | Foarte frecvente (25,6) | 0,7 | Frecvente (5,5) | 0 | Frecvente (6,7) | 0 |
| Durere abdominalㆠ| Foarte frecvente (14,8) | 1,3 | Foarte frecvente (12,1) | 2,0 | Frecvente (7,9) | 1,0 | Frecvente (8,1) | 0,7 |
| Tulburări cutanate și ale țesutului subcutanat | ||||||||
| Erupție cutanată tranzitorie† | Foarte frecvente (30,9) | 1,8 | Foarte frecvente (39,1) | 9,8 | Foarte frecvente (12,0) | 0,3 | Foarte frecvente (16,2) | 1,1 |
| Tulburări musculo-scheletice și ale țesutului conjunctiv | ||||||||
| Durere musculo- scheleticㆠ| Foarte frecvente (44,8) | 2,2 | Foarte frecvente (34,3) | 3,7 | Foarte frecvente (24,1) | 0,7 | Foarte frecvente (21,8) | 1,1 |
| Artralgie | Foarte frecvente (26,9) | 1,3 | Foarte frecvente (17,5) | 0,7 | Foarte frecvente (12,7) | 1,0 | Foarte frecvente (10,9) | 0,4 |
| Tulburări generale și la nivelul locului de administrare | ||||||||
| Fatigabilitate | Foarte frecvente (30,5) | 1,8 | Foarte frecvente (29,3) | 2,7 | Foarte frecvente (14,8) | 0,3 | Foarte frecvente (14,4) | 0 |
| Astenie | Frecvente (7,6) | 0,4 | Foarte frecvente (10,4) | 1,0 | Frecvente (4,1) | 0 | Frecvente (3,2) | 0 |
| Investigații diagnostice¶ | ||||||||
| Scăderea numărului absolut de neutrofile§ | Foarte frecvente (57,4) | 35 | Foarte frecvente (76,8) | 56,6 | Foarte frecvente (78,0) | 38,1 | Foarte frecvente (81,7) | 53,5 |
| Scăderea numărului de trombocite§ | Foarte frecvente (46,2) | 10,8 | Foarte frecvente (69,4) | 17,8 | Foarte frecvente (42,6) | 5,2 | Foarte frecvente (54,9) | 13,7 |
| Scăderea valorilor hemoglobinei§ | Foarte frecvente (43,9) | 9 | Foarte frecvente (79,5) | 10,8 | Foarte frecvente (34,7) | 6,5 | Foarte frecvente (45,8) | 3,5 |
| Creșterea nivelului alanin aminotransferazei‡ | - | - | Frecvente (9,1) | 4,4 | - | - | - | - |
| Creșterea nivelului aspartat aminotransferazei‡ | - | - | Frecvente (8,1) | 3,0 | - | - | - | - |
*Conform Criteriilor de terminologie comună pentru evenimentele adverse ale Institutului Național Oncologic (National Cancer Institute Common Terminology Criteria for Adverse Events, NCI CTCAE), versiunea 4.03. †Include mai mulți termeni pentru RAIM.
±A fost raportat un eveniment advers care a determinat decesul pacientului.
§Reprezintă incidența valorilor rezultatelor de laborator, nu a evenimentelor adverse raportate.
¶Prezentate pe grade de severitate CTCAE.
‡Reacție adversă raportată doar la grupul de pacienți tratați cu Calquence + BR din studiul ECHO.
Descrierea reacțiilor adverse selectate
Infecții grave raportate la pacienții tratați cu Calquence în asociere cu venetoclax cu sau fără obinutuzumab
În rândul celor 291 de pacienți tratați cu Calquence în asociere cu venetoclax, infecțiile severe (grad ≥ 3) au fost raportate la 12,4% dintre pacienți (cel mai frecvent raportate au fost COVID-19 sau pneumonia asociată COVID-19). Infecțiile care au determinat deces au fost raportate la 3,1% dintre pacienți (cel mai frecvent raportate au fost COVID-19 sau pneumonia asociată COVID-19).
În rândul celor 284 de pacienți tratați cu Calquence în asociere cu venetoclax și obinutuzumab, infecțiile grave (grad ≥ 3) au fost raportate la 23,6% dintre pacienți (cel mai frecvent raportate au fost COVID-19 sau pneumonia asociată COVID-19). Infecțiile care au determinat deces au fost raportate la 5,6% dintre pacienți (cel mai frecvent raportate au fost COVID-19 sau pneumonia asociată COVID-19).
Întreruperea administrării şi reducerea dozei din cauza reacţiilor adverse
În rândul celor 1478 de pacienţi trataţi cu Calquence în monoterapie, cazurile de întrerupere a tratamentului din cauza reacţiilor adverse au fost raportate la 14,6% dintre pacienţi. Principalele reacţii adverse au fost pneumonia, trombocitopenia şi diareea. Reducerile dozei din cauza reacţiilor adverse au fost raportate la 5,9% dintre pacienţi. Principalele reacţii adverse au fost reactivarea hepatitei B, sepsisul şi diareea.
În rândul celor 223 de pacienţi trataţi cu Calquence în asociere cu obinutuzumab, cazurile de întrerupere a tratamentului cu Calquence din cauza reacţiilor adverse au fost raportate la 10,8% dintre pacienţi. Principalele reacţii adverse au fost pneumonia, trombocitopenia şi diareea. Reducerile dozei din cauza reacţiilor adverse au fost raportate la 6,7% dintre pacienţi. Principalele reacţii adverse au fost neutropenia, diareea şi vărsăturile.
În rândul celor 291 de pacienți tratați cu Calquence în asociere cu venetoclax, cazurile de întrerupere definitivă a tratamentului cu Calquence din cauza reacțiilor adverse au fost raportate la 7,6% dintre pacienți și cazurile de reducere a dozelor de Calquence din cauza reacțiilor adverse au fost raportate la 5,8% dintre pacienți. Principalele reacții adverse care au determinat întreruperea definitivă a tratamentului au inclus pneumonia asociată COVID-19 și COVID-19, iar reacția adversă care a determinat reducerea dozelor a fost neutropenia.
În rândul celor 284 de pacienți tratați cu Calquence în asociere cu venetoclax și obinutuzumab, cazurile de întrerupere definitivă a tratamentului cu Calquence din cauza reacțiilor adverse au fost raportate la 13,7% dintre pacienți și cazurile de reducere a dozelor de Calquence din cauza reacțiilor adverse au fost raportate la 6,3% dintre pacienți. Principalele reacții adverse care au determinat întreruperea definitivă a tratamentului au inclus pneumonia asociată COVID-19 și COVID-19, iar reacția adversă care a determinat reducerea dozelor a fost neutropenia.
În rândul celor 297 de pacienți tratați cu Calquence în asociere cu bendamustină și rituximab, cazurile de întrerupere definitivă a tratamentului cu Calquence din cauza reacțiilor adverse au fost raportate la 42,8% dintre pacienți. Principalele reacții adverse au fost COVID-19, pneumonie asociată COVID-19, neutropenie și pneumonie. Reducerile dozei din cauza reacțiilor adverse au fost raportate la 10,1% dintre pacienți. Principalele reacții adverse au inclus neutropenie și greață.
Vârstnici
Dintre cei 1478 de pacienţi din studiile clinice cu Calquence în monoterapie, 42% au avut vârsta peste 65 de ani şi sub 75 de ani şi 20,6% au avut vârsta de 75 de ani sau peste. Nu au fost observate diferenţe relevante clinic între pacienţii cu vârsta ≥ 65 ani şi pacienţii mai tineri în ceea ce priveşte siguranţa sau eficacitatea tratamentului.
Dintre cei 223 de pacienţi din studiile clinice cu Calquence ca terapie asociată cu obinutuzumab, 47% au avut vârsta peste 65 de ani şi sub 75 de ani şi 26% au avut vârsta de 75 de ani sau peste. Nu au fost observate diferenţe relevante clinic între pacienţii cu vârsta ≥ 65 ani şi pacienţii mai tineri în ceea ce priveşte siguranţa sau eficacitatea tratamentului.
Dintre cei 291 de pacienți tratați cu Calquence în asociere cu venetoclax, 28,9% au avut vârsta peste 65 de ani și sub 75 de ani și 4,5% au avut vârsta de 75 de ani sau peste. Nu au fost observate diferențe relevante clinic între pacienții cu vârsta ≥ 65 de ani și pacienții mai tineri, în ceea ce privește siguranța și eficacitatea tratamentului.
Dintre cei 284 de pacienți tratați cu Calquence în asociere cu venetoclax și obinutuzumab, 24% au avut vârsta peste 65 de ani și sub 75 de ani și 6,3% au avut vârsta de 75 de ani sau peste. Nu au fost observate diferențe relevante clinic între pacienții cu vârsta ≥ 65 de ani și pacienții mai tineri, în ceea ce privește siguranța și eficacitatea tratamentului.
Raportarea reacţiilor adverse suspectate
Raportarea reacțiilor adverse suspectate după autorizarea medicamentului este importantă. Acest lucru permite monitorizarea continuă a raportului beneficiu/risc al medicamentului. Profesioniştii din domeniul sănătăţii sunt rugaţi să raporteze orice reacţie adversă suspectată prin intermediul sistemului naţional de raportare, astfel cum este menţionat în Anexa V.
4.9 Supradozaj
Nu există un tratament specific pentru supradozajul cu acalabrutinib şi simptomele supradozajului nu au fost stabilite. În caz de supradozaj, pacienţii trebuie atent monitorizaţi pentru apariţia semnelor sau simptomelor de reacţii adverse şi trebuie iniţiat tratamentul simptomatic corespunzător.
5. PROPRIETĂŢI FARMACOLOGICE
5.1 Proprietăţi farmacodinamice
Grupa farmacoterapeutică: Medicamente antineoplazice, inhibitori de protein-kinază, cod ATC: L01EL02.
Mecanism de acţiune
Acalabrutinib este un inhibitor selectiv al tirozin kinazei Bruton (Bruton tyrosine kinase, TKB). TKB este o moleculă de semnalizare a căilor receptorului pentru antigen al celulelor B (B-cell antigen receptor, BCR) şi ale receptorilor citokinici. Semnalizarea TKB la nivelul celulelor B are ca rezultat supravieţuirea şi proliferarea acestora şi este necesară pentru adeziunea, traficul şi chemotaxia celulelor.
Acalabrutinib şi metabolitul său activ, ACP-5862, formează o legătură covalentă cu un rest de cisteină din situsul activ al TKB, determinând inactivarea ireversibilă a TKB cu interacţiuni minimale în afara țintei (off target).
Efecte farmacodinamice
La pacienţii cu malignităţi de celulă B cărora li s-a administrat acalabrutinib în doză de 100 mg de două ori pe zi, gradul median de ocupare a TKB la starea de echilibru ≥ 95% în sângele periferic a fost menţinut timp de 12 ore, având ca rezultat inactivarea TKB pe parcursul intervalului recomandat între administrările dozelor.
Electrofiziologia cardiacă
Efectul acalabrutinib asupra intervalului QTc a fost evaluat pe 46 de subiecţi sănătoşi de sex masculin şi feminin în cadrul unui studiu clinic, randomizat, dublu-orb, asupra intervalului QT controlat cu placebo şi comparator activ. La administrarea în doză supraterapeutică, de 4 ori mai mare decât doza maximă recomandată, Calquence nu a prelungit intervalul QT/QTc într-o măsură relevantă clinic (de exemplu, ≥ 10 ms) (vezi pct. 4.4, 4.8 şi 5.3).
Eficacitate și siguranță clinică
Pacienţii cu LLC netratată anterior
Calquence în monoterapie sau în asociere cu obinutuzumab
Siguranţa şi eficacitatea Calquence în monoterapie sau în asociere cu obinutuzumab în LLC netratată anterior a fost evaluată într-un studiu randomizat, multicentric, deschis, de fază 3 (ELEVATE-TN) la 535 de pacienţi. Pacienţilor li s-a administrat Calquence în asociere cu obinutuzumab, Calquence în monoterapie sau obinutuzumab în asociere cu clorambucil. Studiul ELEVATE-TN a inclus pacienţi cu vârsta de 65 de ani ori peste sau cu vârsta între 18 şi 65 de ani şi afecţiuni medicale concomitente, iar 27,9% dintre pacienţi au avut valori ale ClCr < 60 ml/min. Dintre pacienţii cu vârsta < 65 de ani, 16,1% au avut un scor CIRS-G median de 8. Pacienţilor li s-a permis să administreze medicamente antitrombotice în cadrul studiului. Pacienţii care necesitau tratament anticoagulant cu warfarină sau antagonişti ai vitaminei K echivalenţi au fost excluşi.
Pacienţii au fost randomizaţi în raport de 1:1:1 în 3 braţe de tratament pentru a li se administra
- Calquence în asociere cu obinutuzumab (Calquence+G): Calquence în doză de 100 mg a fost administrat de două ori pe zi începând din ziua 1 a ciclului 1, până la progresia bolii sau apariţia toxicităţii inacceptabile. Obinutuzumab a fost administrat începând din ziua 1 a ciclului 2 timp de maximum 6 cicluri de tratament. Obinutuzumab 1000 mg a fost administrat în zilele 1 şi 2 (100 mg în ziua 1 şi 900 mg în ziua 2), 8 şi 15 ale ciclului 2, iar ulterior în doză de 1000 mg în ziua 1 a ciclurilor 3-7. Fiecare ciclu a avut 28 de zile.
- Calquence în monoterapie: Calquence în doză de 100 mg a fost administrat de două ori pe zi până la progresia bolii sau apariţia toxicităţii inacceptabile.
- Obinutuzumab în asociere cu clorambucil (GClb): Obinutuzumab şi clorambucil au fost administrate timp de maximum 6 cicluri de tratament. Obinutuzumab 1000 mg a fost administrat în zilele 1 şi 2 (100 mg în ziua 1 şi 900 mg în ziua 2), 8 şi 15 ale ciclului 1, iar ulterior în doză de 1000 mg în ziua 1 a ciclurilor 2-6. Clorambucil în doză de 0,5 mg/kg a fost administrat în zilele 1 şi 15 ale ciclurilor 1-6. Fiecare ciclu a avut 28 de zile.
Pacienţii au fost stratificaţi în funcţie de statusul deleţiei 17p (prezenţă versus absenţă), statusul de performanţă ECOG (scor 0 sau 1 versus 2) şi regiunea geografică (America de Nord şi Europa Occidentală versus alte regiuni). După progresia confirmată a bolii, 45 de pacienţi randomizaţi în braţul cu GClb au trecut la tratamentul cu Calquence în monoterapie. Tabelul 6 prezintă rezumativ caracteristicile demografice şi caracteristicile bolii pentru populaţia de pacienţi a studiului la momentul iniţial.
Tabelul 6. Caracteristicile iniţiale ale pacienţilor (ELEVATE-TN) cu LLC netratată anterior
| Caracteristică | Calquence în asociere cu obinutuzumab N=179 | Calquence în monoterapie N=179 | Obinutuzumab în asociere cu clorambucil N=177 |
|---|---|---|---|
| Vârsta în ani; valoare mediană (interval) | 70 (41-88) | 70 (44-87) | 71 (46-91) |
| Sex masculin; % | 62 | 62 | 59,9 |
| Rasă caucaziană; % | 91,6 | 95 | 93,2 |
| Status de performanță ECOG 0-1; % | 94,4 | 92,2 | 94,4 |
| Intervalul median de timp de la diagnostic (luni) | 30,5 | 24,4 | 30,7 |
| Ganglioni voluminoşi cu dimensiunea ≥ 5 cm; % | 25,7 | 38 | 31,1 |
Categoria citogenetică/FISH; % Deleţia 17p Deleţia 11q Mutaţia TP53 IGHV fără mutaţii Cariotip complex (≥ 3 anomalii) | 9,5 17,3 11,7 57,5 16,2 | 8,9 17,3 10,6 66,5 17,3 | 9 18,6 11,9 65,5 18,1 |
| Stadiu Rai; % | |||
0 I II III IV | 1,7 30,2 20,1 26,8 21,2 | 0 26,8 24,6 27,9 20,7 | 0,6 28,2 27,1 22,6 21,5 |
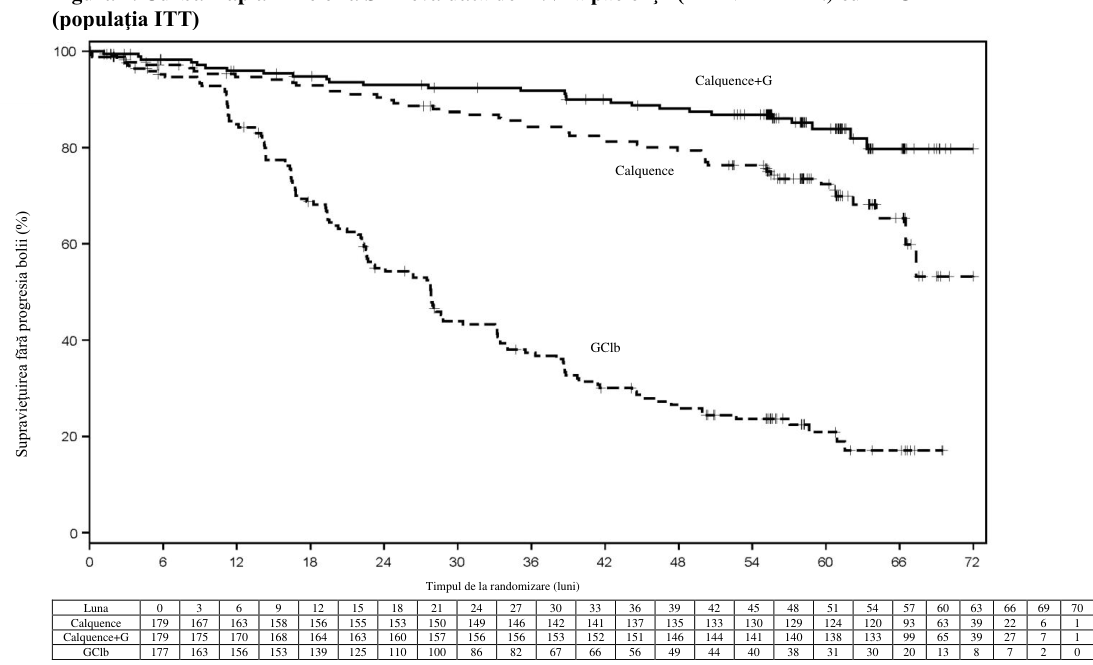
Obiectivul principal a fost supraviețuirea fără progresia bolii (SFP) în braţul de tratament cu Calquence+G comparativ cu braţul tratat cu GClb, evaluată de o comisie independentă de evaluare (IRC) pe baza criteriilor din 2008 ale Grupului internaţional de lucru asupra leucemiei limfocitare cronice (International Workshop on Chronic Lymphocytic Leukaemia, IWCLL), cu includerea clarificării referitoare la limfocitoza asociată tratamentului (Cheson 2012). După o perioadă mediană de urmărire de 28,3 luni, SFP evaluată de IRC a indicat o reducere cu 90%, semnificativă statistic, a riscului de progresie a bolii sau deces pentru pacienţii cu LLC netratată anterior din braţul cu Calquence+G, comparativ cu pacienţii braţului cu GClb. Rezultatele cu privire la eficacitate sunt prezentate în tabelul 7.
Tabelul 7. Rezultatele privind eficacitatea conform evaluărilor IRC (ELEVATE-TN) la pacienţi cu
LLC
| Calquence în asociere cu obinutuzumab N=179 | Calquence în monoterapie N=179 | Obinutuzumab în asociere cu clorambucil N=177 | |
| Supraviețuirea fără progresia bolii* | |||
|---|---|---|---|
| Număr de evenimente (%) | 14 (7,8) | 26 (14,5) | 93 (52,5) |
| PB, n (%) | 9 (5) | 20 (11,2) | 82 (46,3) |
| Număr de decese (%) | 5 (2,8) | 6 (3,4) | 11 (6,2) |
| Valoare mediană (IÎ 95%), luni | NA | NA (34,2, NA) | 22,6 (20,2, 27,6) |
| RR† (IÎ 95%) | 0,10 (0,06, 0,17) | 0,20 (0,13, 0,30) | - |
| Valoare p | <0,0001 | <0,0001 | - |
| Estimare la 24 de luni, % (IÎ 95%) | 92,7 (87,4, 95,8) | 87,3 (80,9, 91,7) | 46,7 (38,5, 54,6) |
| Supravieţuirea generalăa | |||
| Număr de decese (%) | 9 (5) | 11 (6,1) | 17 (9,6) |
| Risc relativ (IÎ 95%) † | 0,47 (0,21, 1,06) | 0,60 (0,28, 1,27) | - |
| Rata celui mai bun răspuns general* (RC + RCi + RPn + RP) | |||
| RRG, n (%) (IÎ 95%) | 168 (93,9) (89,3; 96,5) | 153 (85,5) (79,6; 89,9) | 139 (78,5) (71,9; 83,9) |
| Valoare p | <0,0001 | 0,0763 | - |
| RC, n (%) | 23 (12,8) | 1 (0,6) | 8 (4,5) |
| RCi, n (%) | 1 (0,6) | 0 | 0 |
| RPn, n (%) | 1 (0,6) | 2 (1,1) | 3 (1,7) |
| RP, n (%) | 143 (79,9) | 150 (83,8) | 128 (72,3) |
IÎ = interval de încredere; RR = risc relativ; NA = nu a fost atinsă; RC = răspuns complet; RCi = răspuns complet cu recuperare hematologică incompletă; RPn = răspuns parţial nodular; RP = răspuns parţial;
*Conform evaluării de către IRC.
†Pe baza modelului stratificat Cox al riscurilor proporţionale. a Mediana SG nu a fost atinsă în ambele braţe de tratament.
Rezultatele privind SFP pentru Calquence în asociere cu sau fără obinutuzumab au fost consecvente la nivelul subgrupurilor, inclusiv al celor cu factori de risc înalt. La nivelul populaţiei de pacienţi cu LLC cu risc înalt (deleţie 17p, deleţie 11q, mutaţie TP53 sau IGHV fără mutaţii), RR aferent SFP pentru Calquence asociat sau nu cu obinutuzumab comparativ cu obinutuzumab plus clorambucil a fost de 0,08 [IÎ 95% (0,04, 0,15)] şi, respectiv, de 0,13 [IÎ 95% (0,08, 0,21)].
Tabelul 8. Analiza pe subgrupuri a SFP (studiul ELEVATE-TN)
| Calquence în monoterapie | Calquence+G | |||||
| N | Risc relativ | IÎ 95% | N | Risc relativ | IÎ 95% | |
| Toţi subiecţii | 179 | 0,20 | (0,13; 0,30) | 179 | 0,10 | (0,06; 0,17) |
Del 17p Da Nu | 19 160 | 0,20 0,20 | (0,06; 0,64) (0,12; 0,31) | 21 158 | 0,13 0,09 | (0,04; 0,46) (0,05; 0,17) |
Mutaţia TP53 Da Nu | 19 160 | 0,15 0,20 | (0,05; 0,46) (0,12; 0,32) | 21 158 | 0,04 0,11 | (0,01; 0,22) (0,06; 0,20) |
Del 17p sau/şi mutaţia TP53 Da Nu | 23 156 | 0,23 0,19 | (0,09; 0,61) (0,11; 0,31) | 25 154 | 0,10 0,10 | (0,03; 0,34) (0,05; 0,18) |
Status IGHV Cu mutaţii Fără mutaţii | 58 119 | 0,69 0,11 | (0,31; 1,56) (0,07; 0,19) | 74 103 | 0,15 0,08 | (0,04; 0,52) (0,04; 0,16) |
Del 11q Da Nu | 31 148 | 0,07 0,26 | (0,02; 0,22) (0,16; 0,41) | 31 148 | 0,09 0,10 | (0,03; 0,26) (0,05; 0,20) |
Cariotip complex Da Nu | 31 117 | 0,10 0,27 | (0,03; 0,33) (0,16; 0,46) | 29 126 | 0,09 0,11 | (0,03; 0,29) (0,05; 0,21) |
Conform datelor pe durată îndelungată, perioada mediană de urmărire a fost de 58,2 luni pentru brațul de tratament cu Calquence+G, 58,1 luni pentru brațul de tratament cu Calquence și 58,2 luni pentru brațul de tratament cu GClb. Mediana SFP evaluată de investigator nu a fost atinsă în braţele de tratament cu Calquence+G și Calquence în monoterapie şi a fost de 27,8 luni în braţul cu GClb. La momentul celei mai recente întreruperi a colectării datelor, un total de 72 de pacienți (40,7%) inițial randomizați în brațul cu GClb au trecut la tratamentul cu Calquence în monoterapie. Mediana supraviețuirii generale nu a fost atinsă în niciun braţ de tratament cu un total de 76 de decese: 18 (10,1%) în brațul cu Calquence+G, 30 (16,8%) în brațul cu Calquence în monoterapie și 28 (15,8%) în brațul cu GClb.
Tabelul 9. Rezultatele privind eficacitatea conform evaluărilor INV (ELEVATE-TN) la pacienţi cu
LLC
| Calquence în asociere cu obinutuzumab N=179 | Calquence în monoterapie N=179 | Obinutuzumab în asociere cu clorambucil N=177 | |
| Supraviețuirea fără progresia bolii | |||
|---|---|---|---|
| Număr de evenimente (%) | 27 (15,1) | 50 (27,9) | 124 (70,1) |
| PB, n (%) | 14 (7,8) | 30 (16,8) | 112 (63,3) |
| Număr de decese (%) | 13 (7,3) | 20 (11,2) | 12 (6,8) |
| Valoare mediană (IÎ 95%), luni* | NA | NA (66,5, NA) | 27,8 (22,6, 33,2) |
| RR† (IÎ 95%) | 0,11 (0,07, 0,16) | 0,21 (0,15, 0,30) | - |
| Supraviețuirea generală | |||
| Număr de decese (%) | 18 (10,1) | 30 (16,8) | 28 (15,8) |
| Risc relativ (IÎ 95%)† | 0,55 (0,30, 0,99) | 0,98 (0,58, 1,64) | - |
IÎ=interval de încredere; RR=risc relativ; NA=nu a fost atinsă
*95% interval de încredere bazat pe estimarea Kaplan-Meier.
†Estimare bazată pe modelul stratificat Cox al riscurilor proporţionale pentru riscul relativ (IÎ 95%) stratificat în funcție de statusul deleției 17p (da sau nu).
Figura 1. Curba Kaplan-Meier a SFP evaluată de INV la pacienţii (ELEVATE-TN) cu LLC
Pacienți cu LLC netratată anterior – terapie cu durată fixă
Calquence în asociere cu venetoclax cu sau fără obinutuzumab
Siguranța și eficacitatea Calquence în asociere cu venetoclax cu sau fără obinutuzumab în LLC netratată anterior au fost evaluate într-un studiu randomizat, multicentric, cu design deschis, de fază 3 (AMPLIFY) la 867 pacienți. Pacienților li s-a administrat Calquence în asociere cu venetoclax, Calquence în asociere cu venetoclax și obinutuzumab sau chimioimunoterapie la decizia medicului investigator, fie FCR (fludarabină plus ciclofosfamidă plus rituximab) sau BR (bendamustină plus rituximab). AMPLIFY a inclus pacienți cu LLC netratată anterior, fără mutații de tip deleție (17p) sau TP53, care au avut vârsta de 18 ani și peste. Studiul a permis ca pacienții să primească agenți antitrombotici, mai puțin warfarină sau alți antagoniști ai vitaminei K.
Pacienții au fost randomizați în raport de 1:1:1 în 3 brațe de tratament pentru a li se administra:
- Calquence plus venetoclax (AV): Calquence în doză de 100 mg a fost administrat de două ori pe zi începând cu ziua 1 a primului ciclu de tratament, pentru un număr total de 14 cicluri sau până la progresia bolii sau toxicitate inacceptabilă. În ziua 1 a ciclului 3, pacienții au inițiat programul de titrare a dozelor de venetoclax cu durata de 5 săptămâni, cu doze începând de la 20 mg, care au crescut săptămânal la 50 mg, 100 mg, 200 mg și, la final, 400 mg administrate o dată pe zi. Venetoclax a fost administrat pentru un număr total de 12 cicluri de tratament. Fiecare ciclu a avut 28 de zile.
- Calquence plus venetoclax plus obinutuzumab (AVO): Calquence în doză de 100 mg a fost administrat de două ori pe zi începând cu ziua 1 a primului ciclu de tratament, pentru un număr total de 14 cicluri sau până la progresia bolii sau toxicitate inacceptabilă. În ziua 1 a ciclului 3, pacienții au inițiat programul de titrare a dozelor de venetoclax cu durata de 5 săptămâni, cu doze începând de la 20 mg și care au crescut săptămânal la 50 mg, 100 mg, 200 mg și, la final, 400 mg administrate o dată pe zi. Venetoclax a fost administrat pentru un număr total de 12 cicluri de tratament. Obinutuzumab în doză de 1000 mg a fost administrat în ziua 1 sau în ziua 1 și 2 (100 mg în ziua 1 și 900 mg în ziua 1 sau 2), în zilele 8 și 15 ale ciclului 2, urmat de 1000 mg în ziua 1 a ciclurilor 3-7. Fiecare ciclu a avut 28 de zile.
- Chimioimunoterapie la decizia medicului investigator (FCR/BR):
- Fludarabină plus ciclofosfamidă plus rituximab (FCR): fludarabină (25 mg/m2) și ciclofosfamidă (250 mg/m2) au fost administrate în zilele 1-3, până la un maxim de 6 cicluri de tratament. Rituximab a fost administrat în doză de 375 mg/m2 în ziua 1 a ciclului 1 și 500 mg/m2 în ziua 1 a ciclurilor 2-6. Fiecare ciclu a avut 28 de zile.
- Bendamustină plus rituximab (BR): bendmaustină în doză de 90 mg/m2 a fost administrată în zilele 1 și 2, până la un maxim de 6 cicluri. Rituximab a fost administrat în doză de 375 mg/m2 în ziua 1 a ciclului 1 și 500 mg/m2 în ziua 1 a ciclurilor 2-6. Fiecare ciclu a avut 28 de zile.
Pacienții au fost stratificați în funcție de vârstă (> 65 de ani sau ≤ 65 de ani), de statusul mutațional IGHV (cu mutație sau fără mutație), de stadiul Rai (cu risc crescut [≥ 3] versus fără risc crescut) și de regiunea geografică (America de Nord și Europa de Vest versus altele). Tabelul 10 prezintă sumarul caracteristicilor demografice și clinice la momentul inițial pentru populația de pacienți din studiu.
Tabelul 10. Caracteristicile inițiale ale pacienților (AMPLIFY) cu LLC netratată anterior
| Caracteristică | AV N=291 | AVO N=286 | FCR/BR N=290 |
|---|---|---|---|
| Vârsta în ani; valoare mediană (interval) | 61 (31-84) | 61 (29-81) | 61 (26-86) |
| Sex masculin; % | 61,2 | 69,2 | 63,1 |
| Rasă caucaziană; % | 91,1 | 86,7 | 86,9 |
| Status de performanță ECOG 0-1; % | 90,0 | 95,1 | 90,3 |
| Intervalul median de timp de la diagnostic până la randomizare (luni) | 28,5 | 26,1 | 29,6 |
| Ganglioni voluminoși cu dimensiunea ≥5 cm; % | 38,8 | 35,0 | 42,8 |
| Categoria citogenetică/FISH; % | |||
| Deleție 11q | 17,5 | 19,6 | 15,9 |
| Cariotip complex (≥ 3 anomalii) | 15,5 | 16,1 | 14,5 |
| IGHV fără mutații; % | 57,4 | 59,1 | 59,3 |
| Stadiul Rai; % | |||
| 0 | 1,0 | 0,3 | 1,4 |
| I | 16,2 | 21,3 | 21,4 |
| II | 35,7 | 37,8 | 33,4 |
| III | 23,7 | 17,8 | 20,3 |
| IV | 23,4 | 22,7 | 23,4 |
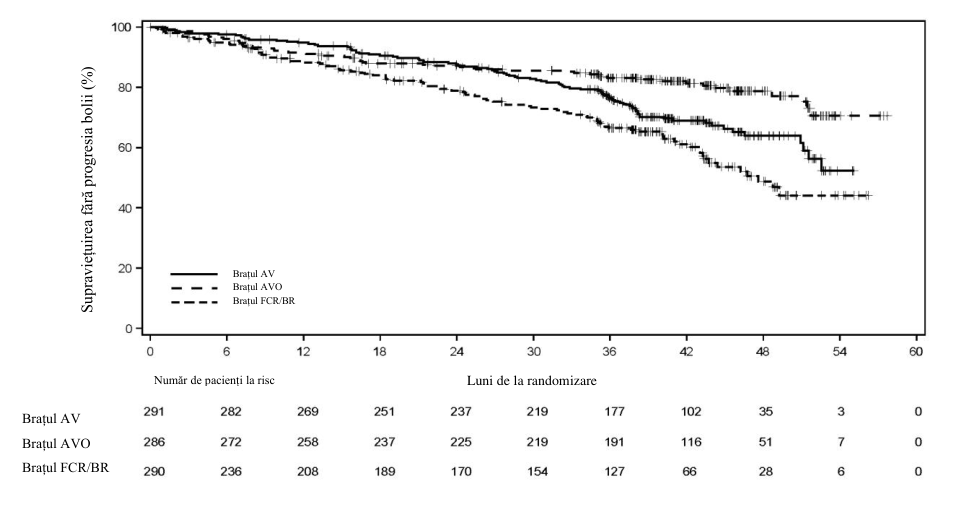
Obiectivul principal a fost SFP în brațul de tratament AV comparativ cu chimioimunoterapia aleasă de medicul investigator (FCR/BR), evaluată conform IRC pe baza criteriilor din 2018 ale IWCLL. Alte obiective ale eficacității tratamentului au fost SFP în brațul de tratament AVO comparativ cu chimioimunoterapie aleasă de medicul investigator (FCR/BR) și SG atât în brațul de tratament AV comparativ cu chimioimunoterapie aleasă de medicul investigator (FCR/BR), cât și în brațul AVO comparativ cu chimioimunoterapie aleasă de medicul investigator (FCR/BR).
Rezultatele privind eficacitatea sunt prezentate în tabelul 11. Curba Kaplan-Meier a SFP-IRC este prezentată în figura 2.
Tabelul 11. Rezultatele privind eficacitatea (AMPLIFY) la pacienții cu LLC netratată anterior
| AV N=291 | AVO N=286 | FCR/BRa N=290 | |
| Supraviețuirea fără progresia bolii* | |||
|---|---|---|---|
| Număr de evenimente (%) | 89 (30,6) | 56 (19,6) | 95 (32,8) |
| PB, n (%) | 77 (26,5) | 23 (8,0) | 66 (22,8) |
| Număr de decese (%) | 12 (4,1) | 33 (11,5) | 29 (10,0) |
| Valoare mediană (IÎ 95%), luni | NC (51,1; NC) | NC (NC; NC) | 47,6 (43,3; NC) |
| RR† (IÎ 95%) | 0,65 (0,49; 0,87) | 0,42 (0,30; 0,59) | - |
| Valoarea p | 0,0038 | ˂0,0001 | - |
| Supraviețuirea globalăb | |||
| Număr de decese (%) | 23 (7,9) | 37 (12,9) | 44 (15,2) |
| RR† (IÎ 95%) | 0,42 (0,25; 0,70)c | 0,75 (0,48; 1,16) | - |
NC= Nu a putut fi calculat; IÎ= Interval de încredere; PB= Progresia bolii
*Conform evaluării IRC.
†Conform modelului stratificat Cox al riscurilor proporționale pentru riscul relativ. aConform alegerii medicului investigator, 143 de pacienți au fost planificați să li se administreze FCR și 147 de pacienți să li se administreze BR. bDate privind SG la o perioadă de monitorizare suplimentară cu durata de 6 luni de la momentul analizei intermediare ale SFP. cValoarea p nu este semnificativă după ajustarea pentru multiplicitate.
Figura 2. Curba Kaplan-Meier a SFP evaluată conform IRC la pacienții (AMPLIFY) cu LLC netratată anterior (populaţia ITT)
Pacienţi cu LLC cărora li s-a administrat cel puțin o terapie anterioară
Siguranţa şi eficacitatea Calquence în LLC recidivată sau refractară au fost evaluate într-un studiu randomizat, multicentric, deschis, de fază 3 (ASCEND) la 310 pacienţi cărora li se administrase minimum o terapie anterioară care nu includea inhibitori ai BCL-2 (limfomului cu celule B 2 mature) sau inhibitori ai receptorului celulelor B. Pacienţilor li s-a administrat Calquence în monoterapie sau terapia selectată de investigator fie idelalisib în asociere cu rituximab sau bendamustină în asociere cu rituximab. Pacienţilor li s-a permis să administreze medicamente antitrombotice în cadrul studiului. Pacienţii care necesitau tratament anticoagulant cu warfarină sau antagonişti ai vitaminei K echivalenţi au fost excluşi.
Pacienţii au fost randomizaţi în raport de 1:1 pentru a li se administra:
- Calquence în doză de 100 mg până la progresia bolii sau apariţia toxicităţii inacceptabile, sau
- La alegerea investigatorului:
- Idelalisib în doză de 150 mg de două ori pe zi în asociere cu rituximab în doză de 375 mg/m2 i.v. în ziua 1 a primului ciclu, apoi în doză de 500 mg/m2 i.v. la fiecare două săptămâni până la 4 doze, iar ulterior la intervale de 4 săptămâni până la 3 doze, administrându-se în total 8 perfuzii.
- Bendamustină în doză de 70 mg/m2 (zilele 1 şi 2 ale fiecărui ciclu de 28 de zile) în asociere cu rituximab (375 mg/m2/500 mg/m2) în ziua 1 a fiecărui ciclu de 28 de zile, timp de până la 6 cicluri.
Pacienţii au fost stratificaţi în funcţie de statusul deleţiei 17p (prezenţă versus absenţă), statusul de performanţă ECOG (scor 0 sau 1 versus 2) şi numărul terapiilor anterioare (1 până la 3 versus ≥ 4). După progresia confirmată a bolii, 35 de pacienţi randomizaţi la terapia aleasă de investigator fie idelalisib cu rituximab sau bendamustină cu rituximab au trecut la tratamentul cu Calquence. Tabelul 12 prezintă rezumativ caracteristicile demografice şi caracteristicile bolii pentru populaţia de pacienţi a studiului la momentul iniţial.
Tabelul 12. Caracteristicile iniţiale ale pacienţilor (ASCEND) cu LLC
| Caracteristică | Calquence în monoterapie N=155 | Terapia aleasă de investigator dintre idelalisib + rituximab şi bendamustină + rituximab N=155 |
|---|---|---|
| Vârsta în ani; valoare mediană (interval) | 68 (32-89) | 67 (34-90) |
| Sex masculin; % | 69,7 | 64,5 |
| Rasă caucaziană; % | 93,5 | 91,0 |
| Status de performanță ECOG; % | ||
| 0 | 37,4 | 35,5 |
| 1 | 50,3 | 51,0 |
| 2 | 12,3 | 13,5 |
| Intervalul median de timp de la diagnostic (luni) | 85,3 | 79,0 |
| Ganglioni voluminoşi cu dimensiunea ≥ 5 cm; % | 49,0 | 48,4 |
Numărul median de terapii anterioare pentru LLC (interval) | 1 (1-8) | 2 (1-10) |
Numărul terapiilor anterioare pentru LLC; % 1 2 3 ≥ 4 | 52,9 25,8 11,0 10,3 | 43,2 29,7 15,5 11,6 |
| Categoria citogenetică/FISH; % | ||
| Deleţia 17p | 18,1 | 13,5 |
| Deleţia 11q | 25,2 | 28,4 |
| Mutaţia TP53 | 25,2 | 21,9 |
| IGHV fără mutaţii | 76,1 | 80,6 |
| Cariotip complex (≥ 3 anomalii) | 32,3 | 29,7 |
| Stadiu Rai; % | ||
| 0 | 1,3 | 2,6 |
| I | 25,2 | 20,6 |
| II | 31,6 | 34,8 |
| III | 13,5 | 11,6 |
| IV | 28,4 | 29,7 |
Obiectivul principal a fost SFP determinată de IRC pe baza criteriilor IWCLL 2008, cu includerea clarificării referitoare la limfocitoza asociată tratamentului (Cheson 2012). După o perioadă de urmărire mediană de 16,1 luni, SFP a indicat o reducere cu 69%, semnificativă statistic, a riscului de deces sau progresie a bolii pentru pacienţii din braţul de tratament cu Calquence. Rezultatele cu privire la eficacitate sunt prezentate în tabelul 13. Curba Kaplan-Meier pentru SFP este prezentată în Figura 3.
Tabelul 13. Rezultatele privind eficacitatea conform evaluărilor IRC (ASCEND) la pacienţi cu LLC
| Calquence în monoterapie N=155 | Terapia aleasă de investigator fie idelalisib + rituximab sau bendamustină + rituximab N=155 | |
| Supraviețuirea fără progresia bolii | ||
|---|---|---|
| Număr de evenimente (%) | 27(17,4) | 68 (43,9) |
| PB, n (%) | 19 (12,3) | 59 (38,1) |
| Număr de decese (%) | 8 (5.2) | 9 (5,8) |
| Valoare mediană (IÎ 95%), luni | NA | 16,5 (14,0, 17,1) |
| RR† (IÎ 95%) | 0,31 (0,20, 0,49) | |
| Valoare p | <0,0001 | |
| Estimare la 15 de luni, % (IÎ 95%) | 82,6 (75,0, 88,1) | 54,9 (45,4, 63,5) |
| Supravieţuirea generalăa | ||
| Număr de decese (%) | 15 (9,7) | 18 (11,6) |
| Risc relativ (IÎ 95%) † | 0,84 (0,42, 1,66) | - |
| Rata celui mai bun răspuns general* (RC + RCi + RPn + RP)** | ||
| RRG, n (%) (IÎ 95%) | 126 (81,3) (74,4, 86,6) | 117 (75,5) (68,1; 81,6) |
| Valoare p | 0,2248 | - |
| RC, n (%) | 0 | 2 (1,3) |
| RP, n (%) | 126 (81,3) | 115 (74,2) |
| Durata răspunsului (DR) | ||
| Valoare mediană (IÎ 95%), luni | NA | 13,6 (11,9, NA) |
IÎ = interval de încredere; RR = risc relativ; NA = nu a fost atinsă; RC = răspuns complet; RCi = răspuns complet cu recuperare hematologică incompletă; RPn = răspuns parţial nodular; RP = răspuns parţial; PB = progresia bolii *Conform evaluării IRC
aMediana SG nu a fost atinsă în ambele braţe de tratament. P<0,6089 pentru SG.
**RCi şi RPn au valori de 0.
†Pe baza modelului stratificat Cox al riscurilor proporţionale
Figura 3. Curba Kaplan-Meier a SFP evaluată de IRC la pacienţii (ASCEND) cu LLC (populaţia
ITT)
| Calquence Terapia aleasă de investigator | |||||||||||||||||||||||||
100
80.
Supravieţuirea fără progresia bolii (%)
60.
40.
20.
0. 0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23
Timpul de la randomizare (luni)
| Număr de pacienți la risc | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Luna | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | 23 |
| Calquence | 155 | 153 | 153 | 149 | 147 | 146 | 145 | 143 | 143 | 139 | 139 | 137 | 118 | 116 | 73 | 61 | 60 | 25 | 21 | 21 | 1 | 1 | 1 | 0 |
| Terapia aleasă de investigator | 155 | 150 | 150 | 146 | 144 | 142 | 136 | 130 | 129 | 112 | 105 | 101 | 82 | 77 | 56 | 44 | 39 | 18 | 10 | 8 | 0 | |||
Rezultatele privind SFP pentru Calquence au fost consecvente la nivelul subgrupurilor, inclusiv al celor cu factori de risc înalt. La nivelul populaţiei cu LLC cu risc înalt (deleţie 17p, deleţie 11q, mutaţie TP53 şi IGHV fără mutaţii), RR pentru SFP a fost de 0,27 [IÎ 95% (0,17, 0,44)].
Tabelul 14. Analiza pe subgrupuri a SFP conform evaluării IRC (studiul ASCEND)
| Calquence în monoterapie | |||
| N | Risc relativ | IÎ 95% | |
| Toţi subiecţii | 155 | 0,30 | (0,19; 0,48) |
Del 17p Da Nu | 28 127 | 0,21 0,33 | (0,07; 0,68) (0,21; 0,54) |
Mutaţia TP53 Da Nu | 39 113 | 0,24 0,33 | (0,11; 0,56) (0,20; 0,57) |
Del 17p sau mutaţie TP53 Da Nu | 45 108 | 0,21 0,36 | (0,09; 0,48) (0,21; 0,61) |
| Status IGHV Cu mutaţii Fără mutaţii | 33 118 | 0,32 0,32 | (0,11; 0,94) (0,19; 0,52) |
Del 11q Da Nu | 39 116 | 0,28 0,31 | (0,11; 0,70) (0,19; 0,53) |
Cariotip complex Da Nu | 50 97 | 0,32 0,23 | (0,16; 0,63) (0,12; 0,44) |
Conform evaluării investigatorului la analiza finală, cu o perioadă mediană de urmărire de 46,5 luni pentru Calquence şi de 45,3 luni pentru IR/BR, a fost observată o reducere cu 72% a riscului de progresie a bolii sau deces pentru pacienții din brațul de tratament cu Calquence. Mediana SFP evaluată de investigator nu a fost atinsă în braţul de tratament cu Calquence şi a fost de 16,8 luni în braţul cu IR/BR. Rezultatele cu privire la eficacitate conform evaluărilor investigatorului (INV) sunt prezentate în tabelul 15. Curba Kaplan-Meier pentru SFP conform evaluării INV este prezentată în Figura 4.
Tabelul 15. Rezultatele privind eficacitatea la analiza finală, conform evaluărilor INV la pacienţii
(ASCEND) cu LLC
| Calquence în monoterapie N=155 | Terapia aleasă de investigator fie idelalisib + rituximab sau bendamustină + rituximab N=155 | |
| Supraviețuirea fără progresia bolii* | ||
|---|---|---|
| Număr de evenimente (%) | 62 (40,0) | 119 (76,8) |
| PB, n (%) | 43 (27,7) | 102 (65,8) |
| Număr de decese (%) | 19 (12,3) | 17 (11,0) |
| Valoare mediană (IÎ 95%), luni | NA | 16,8 (14,1, 22,5) |
| RR† (IÎ 95%) | 0,28 (0,20, 0,38) | |
| Supravieţuirea generalăa | ||
| Număr de decese (%) | 41 (26,5) | 54 (34,8) |
| Risc relativ (IÎ 95%) † | 0,69 (0,46, 1,04) | - |
IÎ = interval de încredere; RR = risc relativ; NA = nu a fost atinsă; PB = progresia bolii
*Conform evaluării INV. aMediana SG nu a fost atinsă în ambele braţe de tratament. P=0,0783 pentru SG.
†Pe baza modelului stratificat Cox al riscurilor proporţionale.
Figura 4. Curba Kaplan-Meier a SFP evaluată de INV la analiza finală, la pacienţii (ASCEND) cu
LLC
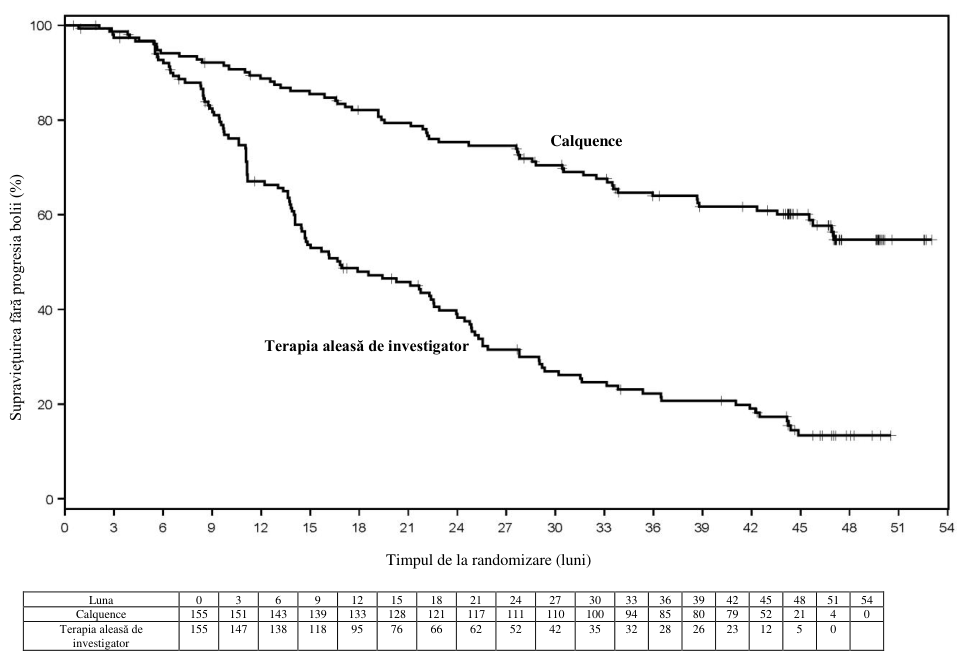
Rezultatele privind SFP pentru Calquence evaluate de investigator la analiza finală au fost consecvente la nivelul subgrupurilor, inclusiv al celor cu factori de risc înalt și au fost în concordanță cu analiza primară.
Pacienți cu LCM netratat anterior
Siguranța și eficacitatea Calquence la pacienții cu LCM fără tratament anterior au fost evaluate în studiul ECHO, un studiu randomizat, de fază III, multicentric, dublu orb, controlat cu placebo. Au fost incluși 598 de pacienți cu vârsta de 65 de ani și peste cu diagnostic confirmat de LCM, fără tratament anterior. Pacienții au fost randomizați în raport 1:1 în două brațe de tratament pentru a li se administra:
- Calquence în asociere cu bendamustină și rituximab (Calquence + BR): s-a administrat Calquence în doze de 100 mg de două ori pe zi în mod continuu începând cu ziua 1 a primului ciclu de tratament. A fost administrată bendamustină în doze de 90 mg/m2 intravenos timp de 30 minute în zilele 1 și 2 ale fiecărui ciclu de tratament cu durata de 28 de zile, pentru un total de șase cicluri; și rituximab în doze de 375 mg/m2 intravenos în ziua 1 a fiecărui ciclu de tratament cu durata de 28 de zile, pentru un total de șase cicluri. Calquence + BR a fost administrat pentru maxim 6 cicluri de tratament (tratament de inducție).
- Placebo în asociere cu bendamustină și rituximab (placebo + BR): s-a administrat placebo de două ori pe zi în mod continuu începând cu ziua 1 a primului ciclu de tratament. A fost administrată bendamustină în doze de 90 mg/m2 intravenos timp de 30 minute în zilele 1 și 2 ale fiecărui ciclu de tratament cu durata de 28 de zile, pentru un total de șase cicluri; și rituximab în doze de 375 mg/m2 intravenos în ziua 1 a fiecărui ciclu de tratament cu durata de 28 de zile, pentru un total de
șase cicluri. Placebo + BR a fost administrat pentru maxim 6 cicluri de tratament (tratament de inducție).
S-a administrat Calquence sau placebo în mod continuu până la progresia bolii sau până la apariția toxicității inacceptabile. După tratamentul de inducție, pacienții care au obținut răspuns (RP sau RC) au primit tratament de menținere cu rituximab în doze de 375 mg/m2 în ziua 1 a fiecărui al doilea ciclu, până la 12 doze suplimentare, până la ciclul 30 de tratament. Pacienții randomizați pentru a li se administra placebo + BR, la care s-a raportat progresia bolii au fost eligibili pentru a face trecerea în brațul cu Calquence ca monoterapie în doze de 100 mg de două ori pe zi, până la a doua progresie a bolii sau până la apariția toxicității inacceptabile.
Procesul de randomizare a pacienților a fost stratificat în funcție de regiunea geografică (America de Nord versus Europa de Vest versus Altele) și de scorul Indicelui de Prognostic Internațional simplificat pentru LCM (Mantle Cell Lymphoma International Prognostic Index, MIPI) (0-3 versus 4-5 versus 6-11).
Vârsta mediană a pacienților a fost de 71 de ani (65-86), 70,7 % au fost bărbați, 78,3% au fost caucazieni și 93,1% au avut status de performanță ECOG 0-1. Scorul simplificat MIPI a fost scăzut (0-3) la 33,1% dintre pacienți, intermediar (4-5) la 42,8% dintre pacienți și crescut (6-11) la 24,1% dintre pacienți. În total, 37,7% dintre pacienți au prezentat masă tumorală ≥ 5 cm și 86% au prezentat boală în stadiul IV conform clasificării Ann Arbor. Au fost raportate variante agresive ale LCM, precum formele blastoide și pleomorfice la 7,7% și 5,5% dintre pacienți. În total, 47,8% dintre pacienți au avut scor Ki-67 ≥ 30%. Caracteristicile inițiale au fost similare în cele două brațe de tratament.
Obiectivul primar a fost supraviețuirea fără progresia bolii (SFP), evaluată de IRC conform Clasificării Lugano 2014 pentru limfom non-Hodkin (LNH) la pacienții cu LCM netratat anterior. Suplimentar, a fost evaluată de IRC și rata de răspuns global (RRG).
SFP a fost evaluată de IRC la o perioadă mediană de urmărire de 49,8 luni.
Supraviețuirea globală mediană nu a fost atinsă în niciun braț de tratament, cu o perioadă de urmărire suplimentară de 6 luni de la analiza primară a SFP și urmărire mediană de 63,0 luni. Au fost raportate 218 decese: 105 (35,1%) în grupul tratat cu Calquence + BR și 113 (37,8%) în grupul cu administrare de placebo + BR. Rezultatele de eficacitate sunt prezentate în tabelul 16. Curbele Kaplan-Meier ale SFP sunt prezentate în figura 5.
Tabelul 16. Rezultate privind eficacitatea la pacienții fără tratament anterior pentru LCM din studiul ECHO
| Calquence + BR N=299 | Placebo + BR N=299 | |
| SFP evaluată conform IRC | ||
|---|---|---|
| Mediana (IÎ 95%) | 66,4 (55,1, NE) | 49,6 (36,0, 64,1) |
| RR (IÎ 95%) (stratificată)* | 0,73 (0,57, 0,94) | |
| Valorea p‡ | 0,0160 | |
| RRG evaluată conform IRC | ||
| RC + RP n (%) | 272 (91,0) | 263 (88,0) |
| IÎ 95% | 87,3, 93,8 | 83,9, 91,3 |
| RC n (%) | 199 (66,6) | 160 (53,5) |
| RP n (%) | 73 (24,4) | 103 (34,4) |
| Valoarea p | 0,2196 | - |
RR = risc relativ, RC = răspuns complet, RP = răspuns parțial, NE = neevaluabil
*Stratificat pe baza factorilor de randomizare: regiune geografică (America de Nord, Europa de Vest, altele) și scorul simplificat MIPI (risc scăzut [între 0 și 3], risc intermediar [între 4 și 5], risc crescut [între 6 și 11], datele fiind colectate cu ajutorul IXRS.
Estimat pe baza modelului stratificat Cox al riscurilor proporționale pentru evaluarea ratei de risc (IÎ 95%). ‡Estimat pe baza testului stratificat log-rank pentru valoarea p.
Figura 5. Curba Kaplan-Meier a SFP evaluată de IRC la pacienții cu LCM fără tratament anterior
(ECHO)
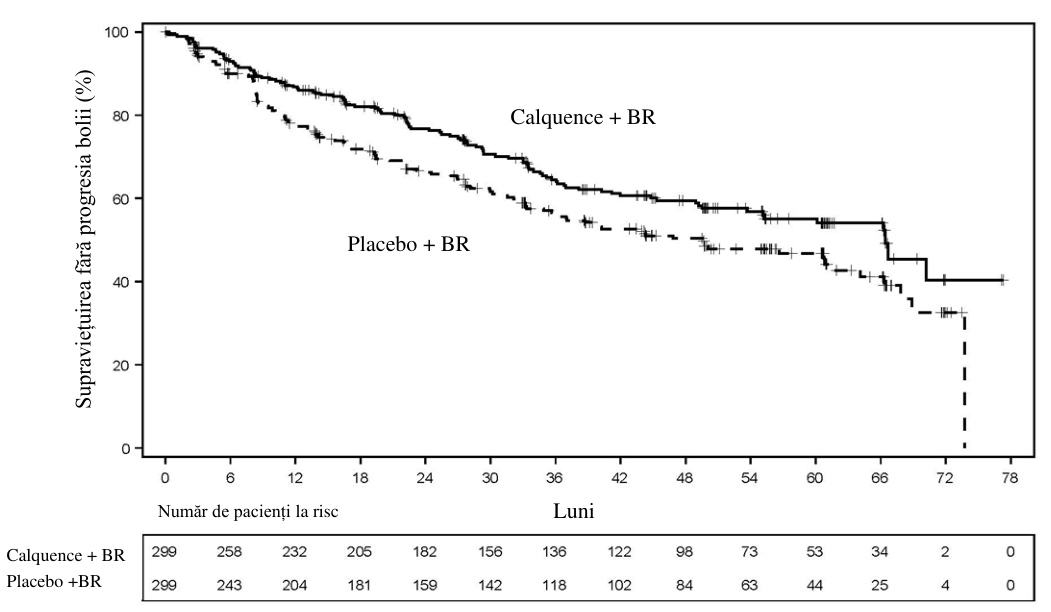
Pacienţi cu LCM cărora li s-a administrat cel puțin o terapie anterioară
Siguranța și eficacitatea CALQUENCE la pacienții cu LCM au fost evaluate în cadrul unui studiu de fază II, deschis, multicentric, cu un singur braț de tratament (ACE-LY-004) care a inclus 124 pacienți tratați anterior. Tuturor pacienţilor li s-a administrat CALQUENCE pe cale orală, în doză de 100 mg de două ori pe zi până la progresia bolii sau toxicitate inacceptabilă. Studiul nu a inclus pacienții cărora li se administrase tratament anterior cu inhibitori TKB sau BCL-2. Obiectivul principal a fost rata răspunsului general (RRG) evaluată de investigator conform clasificării Lugano pentru limfomul non-Hodgkin (non-Hodgkin’s lymphoma, LNH). Un obiectiv suplimentar de evaluare a fost durata răspunsului (DR). Rezultatele eficacității la analiza finală (54 de luni) sunt prezentate în tabelul 17.
La analiza finală, vârsta mediană a fost de 68 de ani (interval 42 – 90 de ani), 79,8% erau bărbați, iar 74,2% erau caucazieni. La momentul inițial, 92,8% dintre pacienți aveau un status al performanței ECOG 0 sau 1. Timpul median de la diagnostic a fost de 46,3 luni iar numărul median de tratamente anterioare a fost de 2 (interval 1 - 5), incluzând cei 17,7% dintre pacienții cu transplant anterior de celule stem. Cele mai frecvente scheme de tratament anterioare au avut la bază regimul CHOP (51,6%) și citarabina
(ARA-C) (33,9%). La momentul inițial, 37,1% dintre pacienți aveau cel puțin o tumoră cu diametrul cel mai mare ≥ 5 cm, iar 72,6% aveau implicare extraganglionară, incluzând cei 50,8% dintre pacienții cu afectare a măduvei osoase. Scorul MIPI simplificat (care include vârsta, scorul ECOG și valorile inițiale de lactat dehidrogenază și ale numărului de leucocite) a fost intermediar la 43,5% dintre pacienți și ridicat la 16,9% dintre pacienți.
Tabelul 17. RRG și DR la pacienții cu LCM din studiul ACE-LY-004 la analiza finală de 54 luni
| Evaluarea investigatorului la 54 luni N=124 n (%) (IÎ 95%*) | |
| Rata răspunsului general (RRG) | |
|---|---|
| Rata răspunsului general | 101 (81,5%) (73,5; 87,9) |
| Răspuns complet | 59 (47,6%) (38,5; 56,7) |
| Răspuns parțial | 42 (33,9%) (25,6; 42,9) |
| Nu poate fi estimatㆠ| 3 (2,4%) (0,5; 6,9) |
| Durata răspunsului (DR) | |
| Valoarea mediană (luni) | 28,6 (17,5; 39,1) |
IÎ=interval de încredere *95% interval de încredere binomial exact. †Include subiecți fără nicio evaluare adecvată a bolii după momentul inițial. | |
Populația pediatrică
Agenţia Europeană a Medicamentului a acordat o derogare de la obligaţia de depunere a rezultatelor studiilor efectuate cu Calquence la toate subgrupele de copii şi adolescenţi pentru tratamentul neoplasmelor cu celule B mature (vezi pct. 4.2 pentru informaţii privind utilizarea la copii şi adolescenţi).
5.2 Proprietăţi farmacocinetice
Farmacocinetica (FC) acalabrutinib şi a metabolitului său activ, ACP-5862, au fost studiate la subiecţi sănătoşi şi la pacienţi cu afecțiuni maligne cu celule B. Expunerea la acalabrutinib este proporţională cu doza şi atât acalabrutinib, cât şi ACP-5862 au o FC aproape liniară la administrarea unor doze cuprinse între 75 şi 250 mg. Modelarea FC populaţională sugerează că farmacocinetica acalabrutinib şi a ACP-5862 este similară la pacienţi cu afecțiuni maligne cu celule B diferite. La doza recomandată de 100 mg de două ori pe zi administrată pacienţilor cu afecțiuni maligne cu celule B (inclusiv LLC), media geometrică a concentraţiilor plasmatice zilnice la starea de echilibru în funcţie de timp (ASC24ore) şi concentraţia plasmatică maximă (Cmax) pentru acalabrutinib au fost de 1679 ngxoră/ml şi, respectiv, de 438 ng/ml, iar pentru ACP-5862 au fost de 4166 ngxoră/ml şi, respectiv, de 446 ng/ml.
Absorbție
Timpul până la atingerea concentraţiilor plasmatice maxime (Tmax) a fost de 0,5-1,5 ore pentru acalabrutinib şi de 1,0 oră pentru ACP-5862. Biodisponibilitatea absolută a Calquence a fost de 25%.
Efectul alimentelor asupra acalabrutinib
La voluntarii sănătoşi, administrarea unei singure doze de 75 mg de acalabrutinib în timpul unei mese hipercalorice şi cu conţinut ridicat de grăsimi (aproximativ 918 calorii, 59 grame carbohidraţi, 59 grame lipide şi 39 grame proteine) nu a influenţat valorile medii ale ASC comparativ cu administrarea în condiţii de repaus alimentar. Valorile Cmax rezultate au scăzut cu 69% şi Tmax a fost decalat cu 1-2 ore.
Distribuție
Proporţia legării reversibile de proteinele plasmatice umane a fost de 99,4% pentru acalabrutinib şi de 98,8% pentru ACP-5862. Raportul mediu sânge-plasmă in vitro a fost de 0,8 pentru acalabrutinib şi de 0,7 pentru ACP-5862. Volumul de distribuţie mediu la starea de echilibru (Vss) a fost de aproximativ 34 l pentru acalabrutinib.
Biotransformare/metabolizare
In vitro, acalabrutinib este predominant metabolizat de enzimele CYP3A şi în proporţie minoră prin conjugarea cu glutation şi hidroliză amidică. ACP-5862 a fost identificat ca metabolit principal în plasmă, care ulterior a fost metabolizat în special prin oxidare mediată de CYP3A, cu o medie geometrică a expunerii (ASC) de aproximativ 2 până la 3 ori mai mare decât expunerea la acalabrutinib. ACP-5862 are o putere de inhibiţie a TKB cu aproximativ 50% mai mică decât cea a acalabrutinibului.
Studiile in vitro indică faptul că acalabrutinib nu inhibă CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, UGT1A1 sau UGT2B7 la concentraţii relevante clinic şi probabilitatea ca acesta să afecteze clearance-ul medicamentelor substrat pentru aceste enzime ale CYP este foarte redusă.
Studiile in vitro indică faptul că ACP-5862 nu inhibă CYP1A2 , CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4/5, UGT1A1 sau UGT2B7 la concentraţii relevante clinic şi probabilitatea ca acesta să afecteze clearance-ul medicamentelor substrat pentru aceste enzime ale CYP este foarte redusă.
Interacţiuni cu proteinele transportoare
Studiile in vitro indică faptul că acalabrutinib şi ACP-5862 sunt substraturi pentru gp-P şi BCRP. Cu toate acestea, este improbabil ca administrarea concomitent cu inhibitori ai BCRP să determine interacţiuni medicamentoase relevante din punct de vedere clinic. Administrarea concomitent cu un inhibitor al OATP1B1/1B3 (rifampicină în doză unică de 600 mg) a determinat o creştere a valorilor Cmax şi ASC ale acalabrutinib de 1,2 ori şi, respectiv, de 1,4 ori (N=24, subiecţi sănătoşi), ceea ce nu este relevant din punct de vedere clinic.
La administrarea în concentraţii relevante clinic, acalabrutinib şi ACP-5862 nu inhibă gp-P, OAT1, OAT3, OCT2, OATP1B1, OATP1B3 şi MATE2-K. Acalabrutinib poate inhiba BCRP de la nivel intestinal, în timp ce ACP-5862 poate inhiba MATE1 la administrarea în concentraţii relevante clinic (vezi pct. 4.5). Acalabrutinib nu inhibă MATE1, iar ACP-5862 nu inhibă BCRP la administrarea în concentraţii relevante clinic.
Eliminare
După administrarea unei singure doze orale de 100 mg de acalabrutinib, timpul terminal de înjumătăţire plasmatică prin eliminare (t1/2) pentru acalabrutinib a fost de 1 până la 2 ore. Pentru metabolitul activ ACP-5862, t1/2 a fost de aproximativ 7 ore.
Valoarea medie a clearance-ului oral aparent (CL/F) a fost de 134 l/oră pentru acalabrutinib şi de 22 l/oră pentru ACP-5862 la toţi pacienţii cu afecțiuni maligne cu celule B.
După administrarea unei singure doze de 100 mg de acalabrutinib marcate radioactiv [14C] la voluntari sănătoşi, 84% din doza administrată a fost recuperată din materiile fecale şi 12% din doză a fost recuperată din urină, mai puţin de 2% din doză fiind excretată sub formă de acalabrutinib nemodificat.
Grupe speciale de pacienți
Pe baza analizei farmacocinetice populaţionale, vârsta (> 18 ani), sexul, rasa (caucaziană, afro-americană) şi greutatea corporală nu au avut efecte relevante clinic asupra farmacocineticii acalabrutinib şi a metabolitului său activ, ACP-5862.
Copii și adolescenți
Nu s-au efectuat studii farmacocinetice cu Calquence la pacienţi cu vârsta sub 18 ani.
Insuficienţă renală
Acalabrutinib este eliminat în proporţie minimală pe cale renală. Nu s-a efectuat niciun studiu farmacocinetic la pacienţi cu insuficiență renală.
Pe baza analizei FC populaţionale, nu au fost observate diferenţe relevante din punct de vedere farmacocinetic între cei 408 de subiecţi cu insuficienţă renală uşoară (RFGe între 60 şi 89 ml/min/1,73 m2, conform estimării prin ecuaţia MDRD), cei 109 subiecţi cu insuficiență renală moderată (RFGe între 30 şi 59 ml/min/1,73 m2) şi cei 192 de subiecţi cu funcţie renală în parametri normali (RFGe mai mare sau egală cu 90 ml/min/1,73 m2). Farmacocinetica acalabrutinib nu a fost caracterizată la pacienţi cu insuficiență renală severă (RFGe sub 29 ml/min/1,73 m2) sau la cei cu insuficiență renală necesitând tratament prin dializă. Pacienţii cu niveluri ale creatininei de 2,5 ori mai mari decât LSVN instituţională nu au fost incluşi în studiile clinice (vezi pct. 4.2).
Insuficiență hepatică
Acalabrutinib este metabolizat pe cale hepatică. În studiile dedicate efectuate la pacienţi cu insuficiență hepatică (IH) comparativ cu pacienţi cu funcţie hepatică normală (N=6), expunerea la acalabrutinib (ASC) a crescut de 1,9 ori, 1,5 ori şi de 5,3 ori la pacienţii cu insuficiență hepatică uşoară (N=6) (Clasificarea Child-Pugh clasa A), moderată (N=6) (Clasificarea Child-Pugh clasa B) şi, respectiv, severă (N=8) (Clasificarea Child-Pugh clasa C). La subiecţii din grupul cu IH moderată, modificările markerilor relevanţi pentru capacitatea de eliminare a medicamentelor nu au fost însă semnificative, prin urmare este posibil ca efectul insuficienţei hepatice moderate să fi fost subestimat în cadrul acestui studiu. Pe baza analizei farmacocinetice populaţionale, nu au fost observate diferenţe relevante clinic între subiecţii cu insuficienţă hepatică uşoară (N=79) sau moderată (N=6) (valori ale bilirubinei totale de 1,5 până la 3 ori mai mari decât LSVN şi orice valori ale AST) şi cei cu funcţie hepatică normală (N=613) (valori ale bilirubinei totale şi AST în limitele LSVN) (vezi pct. 4.2).
5.3 Date preclinice de siguranţă
Carcinogenicitate
Nu s-au efectuat studii de carcinogenicitate cu acalabrutinib.
Genotoxicitate/mutagenicitate/fototoxicitate
Acalabrutinib nu a fost mutagen în cadrul testului mutaţiei inverse bacteriene, al testului in vitro pentru detectarea aberaţiilor cromozomiale sau al testului in vivo al micronucleilor de la nivelul măduvei hematogene la şoarece.
Pe baza testelor de fototoxicitate in vitro pe linii celulare 3T3, se consideră că acalabrutinib prezintă un risc scăzut de fototoxicitate la om.
Toxicitatea după doze repetate
La şobolani au fost observate modificări microscopice la nivelul pancreasului, minimale până la uşoare ca severitate (hemoragie/pigmenţi/inflamaţie/fibroză în insulele pancreatice) la toate dozele testate. Au fost observate modificări non-adverse, minimale până la uşoare ca severitate, la nivelul rinichilor (bazofilie tubulară, regenerare tubulară şi inflamaţie) în studii cu durata de până la 6 luni în care s-a administrat la şobolani doza maximă fără efecte adverse observabile (No Observed Adverse Effect Level, NOAEL) de 30 mg/kg şi zi. Expunerile medii (ASC) în cazul administrării NOAEL la şobolani masculi şi femele corespund unui nivel de 0,6 ori şi respectiv 1,0 ori expunerea clinică la doza recomandată de 100 mg de două ori pe zi. Doza minimă cu efecte adverse observabile (Lowest Adverse Observed Effect Level, LOAEL) la care au fost înregistrate modificări reversibile la nivel renal (degenerare tubulară moderată) şi hepatic (necroza hepatocitelor individuale) în studiul pe termen lung la şobolani a fost de 100 mg/kg şi zi, şi a determinat o expunere de 4,2 ori mai mare decât expunerea cu doza recomandată de 100 mg de două ori pe zi. În studii cu durata de 9 luni efectuate la câini, NOAEL a fost de 10 mg/kg şi zi, ceea ce corespunde unei expuneri de 3 ori mai mare decât ASC la doza clinică recomandată. La administrarea unei doze de 30 mg/kg şi zi la câini (de 9 ori mai mare decât ASC clinică) au fost observate degenerări tubulare minimale la nivelul rinichilor, scăderi uşoare ale greutăţii splinei, scăderi tranzitorii, minimale până la uşoare, ale masei eritrocitare şi creşteri ale ALT şi ALP. Toxicităţile cardiace la şobolani (hemoragie, inflamație, necroză miocardică) şi câini (inflamaţie perivasculară/vasculară) au fost înregistrate numai la animale decedate pe parcursul studiilor, în contextul administrării de doze mai mari decât doza maximă tolerată (DMT). Expunerile la şobolani şi câini asociate cu modificări cardiace au fost cel puţin de 6,8 ori şi, respectiv, de 25 ori mai mari decât ASC clinică. Reversibilitatea acestor modificări cardiace nu a putut fi evaluată deoarece acestea au fost observate numai la administrarea unor doze peste DMT.
Efectele toxice asupra funcţiei de reproducere
Nu au fost observate efecte asupra fertilităţii la şobolanii masculi sau femele la expuneri de 10 sau, respectiv, 9 ori mai mari decât ASC clinică la doza recomandată.
Nu au fost observate efecte asupra dezvoltării şi supravieţuirii embrio-fetale la femelele de şobolan gestante, în cazul unor expuneri de aproximativ 9 ori mai mari decât ASC observată la pacienţii trataţi cu doza recomandată de 100 mg de două ori pe zi. În două studii de evaluare a efectelor asupra reproducerii la şobolani, au fost observate cazuri de distocie (travaliu dificil/prelungit) la expuneri de >2,3 ori mai mari decât expunerea clinică la doza de 100 mg de două ori pe zi. S-a confirmat prezenţa acalabrutinib şi a metabolitului său activ în plasma fetuşilor de şobolan. Prezenţa acalabrutinib şi a metabolitului său activ a fost depistată în laptele matern al femelelor de şobolan.
Într-un studiu de toxicitate embrio-fetală pe femele de iepure gestante au fost observate scăderi ale greutăţii fetuşilor şi întârzieri ale osificării la niveluri de expunere asociate cu toxicitate maternă care au fost de 2,4 ori mai mari decât valorile ASC la om atunci când s-a administrat doza recomandată.
6. PROPRIETĂŢI FARMACEUTICE
6.1 Lista excipienţilor
Conţinutul capsulei
Celuloză microcristalină
Dioxid de siliciu coloidal anhidru
Amidon de porumb parţial pregelatinizat
Stearat de magneziu (E470b)
Amidonglicolat de sodiu
Învelişul capsulei
Gelatină
Dioxid de titan (E171)
Oxid galben de fer (E172)
Carmin indigo (E132)
Cerneală de inscripţionare
Shellac
Oxid negru de fer (E172)
Propilenglicol (E1520)
Hidroxid de amoniu
6.2 Incompatibilităţi
Nu este cazul.
6.3 Perioada de valabilitate
3 ani.
6.4 Precauţii speciale pentru păstrare
Acest medicament nu necesită condiţii speciale de păstrare.
6.5 Natura şi conţinutul ambalajului
Blistere din aluminiu/aluminiu, cu simbolurile soarelui/lunii, care conţin 6 sau 8 capsule. Cutii a câte 56 sau 60 de capsule.
Este posibil ca nu toate mărimile de ambalaj să fie comercializate.
6.6 Precauţii speciale pentru eliminare
Orice medicament neutilizat sau material rezidual trebuie eliminat în conformitate cu reglementările locale.
7. DEŢINĂTORUL AUTORIZAŢIEI DE PUNERE PE PIAŢÃ
AstraZeneca AB
SE-151 85 Södertälje
Suedia
8. NUMĂRUL(ELE) AUTORIZAŢIEI DE PUNERE PE PIAŢĂ
EU/1/20/1479/001
EU/1/20/1479/002
9. DATA PRIMEI AUTORIZĂRI SAU A REÎNNOIRII AUTORIZAŢIEI
Data primei autorizări: 05 noiembrie 2020
Data ultimei reînnoiri a autorizației:
10. DATA REVIZUIRII TEXTULUI
Informaţii detaliate privind acest medicament sunt disponibile pe site-ul Agenţiei Europene pentru Medicamente http://www.ema.europa.eu.
1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI
Calquence 100 mg comprimate filmate
2. COMPOZIŢIA CALITATIVĂ ŞI CANTITATIVĂ
Fiecare comprimat filmat conţine acalabrutinib 100 mg (sub formă de acalabrutinib maleat).
Pentru lista tuturor excipienţilor, vezi pct. 6.1.
3. FORMA FARMACEUTICĂ
Comprimat filmat (comprimat).
Comprimate ovale, biconvexe, de culoare portocalie, cu dimensiuni de 7,5 x 13 mm, marcate cu ‘ACA 100’ pe o față și netede pe cealaltă față.
4. DATE CLINICE
4.1 Indicaţii terapeutice
Calquence în monoterapie sau în asociere cu obinutuzumab este indicat pentru tratamentul pacienţilor adulţi cu leucemie limfocitară cronică (LLC) netratată anterior.
Calquence în asociere cu venetoclax cu sau fără obinutuzumab este indicat pentru tratamentul pacienților adulți cu leucemie limfocitară cronică (LLC) netratată anterior.
Calquence în monoterapie este indicat pentru tratamentul pacienţilor adulţi cu leucemie limfocitară cronică (LLC) cărora li s-a administrat cel puţin o terapie anterioară.
Calquence în asociere cu bendamustină și rituximab (BR) este indicat pentru tratamentul pacienților adulți cu limfom cu celule de manta (LCM) netratat anterior și care nu sunt eligibili pentru transplant autolog de celule stem (TACS).
Calquence în monoterapie este indicat pentru tratamentul pacienților adulți cu limfom cu celule de manta (LCM) recidivat sau refractar netratați anterior cu un inhibitor al tirozin kinazei Bruton (TKB).
4.2 Doze şi mod de administrare
Tratamentul cu acest medicament trebuie iniţiat şi supravegheat de un medic cu experienţă în utilizarea terapiilor antineoplazice.
Doze
Doza recomandată de Calquence în monoterapie sau în asociere cu alte medicamente este de 100 mg de acalabrutinib de două ori pe zi (echivalentul unei doze zilnice totale de 200 mg).
Intervalul de administrare a dozelor de Calquence este de aproximativ 12 ore.
În cazul administrării schemelor terapeutice în asociere, vă rugăm să consultați rezumatul caracteristicilor produsului al fiecărui medicament pentru informații referitoare la dozele recomandate (pentru detalii privind schemele terapeutice în asociere, vezi pct. 5.1).
Calquence în monoterapie sau în asociere cu obinutuzumab
Tratamentul cu Calquence în monoterapie sau în asociere cu obinutuzumab trebuie continuat până la progresia bolii sau până la apariţia toxicităţii inacceptabile.
Calquence în asociere cu venetoclax cu sau fără obinutuzumab
Tratamentul cu Calquence în asociere cu venetoclax cu sau fără obinutuzumab trebuie continuat până la progresia bolii, până la apariția toxicității inacceptabile sau până la finalizarea a 14 cicluri de tratament (fiecare ciclu are 28 de zile).
Calquence trebuie administrat în ziua 1 a primului ciclu, pentru un număr total de 14 cicluri de tratament. Venetoclax trebuie administrat în ziua 1 a ciclului 3, pentru un număr total de 12 cicluri, inițial în doză de 20 mg, care este crescută săptămânal la 50 mg, 100 mg, 200 mg și, la final, 400 mg.
Atunci când Calquence este administrat în asociere cu venetoclax și obinutuzumab, obinutuzumab trebuie administrat în doză de 100 mg în ziua 1 a ciclului 2, urmat de o doză de 900 mg care poate fi administrată în ziua 1 sau 2. Obinutuzumab trebuie administrat în doză de 1000 mg în zilele 8 și 15 ale ciclului 2, urmat de 1000 mg în ziua 1 începând cu ciclul 3 până la ciclul 7. Obinutuzumab se administrează pentru un număr total de 6 cicluri.
Calquence în asociere cu bendamustină și rituximab
Calquence trebuie administrat în ziua 1 a ciclului 1 de tratament (fiecare ciclu are 28 de zile) până la progresia bolii sau până la apariția toxicității inacceptabile. Bendamustina în doze de 90 mg/m2 se administrează în zilele 1 și 2 ale fiecărui ciclu, pentru un număr total de 6 cicluri de tratament. În ziua 1 a fiecărui ciclu se administrează rituximab în doze de 375 mg/m2, pentru un număr total de 6 cicluri de tratament. Pacienții care au obținut răspuns (răspuns parțial [RP] sau răspuns complet [RC]) după primele 6 cicluri de tratament pot să primească tratament de menținere cu rituximab în doze de 375 mg/m2 în ziua 1 a fiecărui al doilea ciclu și pentru maxim 12 doze suplimentare, începând cu ciclul 8 până la ciclul 30 de tratament.
Ajustarea dozelor
Reacţii adverse
Recomandările privind modificarea dozelor de Calquence în cazul reacţiilor adverse de grad ≥ 3 la pacienții cărora li se administrează Calquence în monoterapie, Calquence în asociere cu obinutuzumab sau Calquence în asociere cu venetoclax cu sau fără obinutuzumab sunt prezentate în tabelul 1.
Recomandările privind modificarea dozelor în cazul reacțiilor adverse de grad ≥ 3 la pacienții cărora li se administrează Calquence în asociere cu bendamustină și rituximab sunt prezentate în tabelul 2.
Tabelul 1. Ajustări recomandate ale dozelor în caz de reacţii adverse*
| Reacţie adversă | Apariţia reacţiei adverse | Modificarea dozei (doza de început = 100 mg la intervale aproximative de 12 ore) |
|---|---|---|
| Trombocitopenie de gradul 3 cu sângerare Trombocitopenie de gradul 4 Neutropenie de gradul 4 care persistă mai mult de 7 zile Toxicităţi non-hematologice de gradul 3 sau mai severe | Prima şi a doua | Se va întrerupe administrarea Calquence. Odată ce toxicitatea s-a remis la gradul 1 sau la valorile iniţiale, se poate relua administrarea Calquence în doze de 100 mg la intervale de aproximativ 12 ore. |
| A treia | Se va întrerupe administrarea Calquence. Odată ce toxicitatea s-a remis la gradul 1 sau la valorile iniţiale, se poate relua administrarea Calquence în doze de 100 mg o dată pe zi. | |
| A patra | Se va întrerupe definitiv tratamentul cu Calquence. |
*Reacţiile adverse au fost clasificate pe grade de severitate conform Criteriilor de terminologie comună pentru evenimentele adverse ale Institutului Naţional Oncologic (National Cancer Institute Common Terminology Criteria for Adverse Events, NCI CTCAE), versiunea 5.0.
Tabelul 2. Recomandări privind ajustarea dozelor în cazul reacțiilor adverse de grad ≥ 3* la pacienții tratați cu Calquence în asociere cu bendamustină și rituximab
| Reacție adversă | Modificarea dozei de bendamustinㆠ| Modificarea dozei de Calquence |
|---|---|---|
| Neutropenie | În cazul neutropeniei de grad 3 sau 4‡: se va întrerupe administrarea bendamustinei. Odată ce toxicitatea s-a remis la grad ≤ 2 sau la valorile inițiale, se poate relua administrarea bendamustinei în doze de 70 mg/m2. Se va întrerupe definitiv administrarea bendamustinei dacă sunt necesare reduceri suplimentare ale dozelor. | Se va întrerupe administrarea Calquence dacă neutropenia de grad 4 durează mai mult de 7 zile. Odată ce toxicitatea s-a remis la grad ≤ 2 sau la valorile inițiale, se poate relua administrarea Calquence cu doza de inițiere (la prima apariție a reacției adverse) sau cu frecvență redusă în doză de 100 mg zilnic, (la a doua sau a treia apariție a reacției adverse).¶ Se va întrerupe definitiv administrarea Calquence la a patra apariție a reacției adverse. |
| Trombocitopenie | În cazul trombocitopeniei de grad 3 sau 4: se va întrerupe administrarea bendamustinei. Odată ce toxicitatea s-a remis la grad ≤ 2 sau la valorile inițiale, se poate relua administrarea | Se va întrerupe administrarea Calquence în cazul apariției trombocitopeniei de grad 3 cu sângerare semnificativă sau trombocitopeniei de grad 4. Odată ce toxicitatea s-a remis la grad ≤ 2 sau la valorile inițiale, se poate relua administrarea Calquence cu doza de inițiere (la prima apariție a reacției adverse) sau cu frecvență redusă în doză de 100 mg zilnic |
| bendamustinei în doze de 70 mg/m2. Se va întrerupe definitiv administrarea bendamustinei dacă sunt necesare reduceri suplimentare ale dozelor. | (la a doua sau a treia apariție a reacției adverse).¶ În cazul apariției celei de-a treia reacții adverse de trombocitopenie cu sângerare semnificativă se va întrerupe definitiv administrarea Calquence. Se va întrerupe definitiv administrarea Calquence la a patra apariție a reacției adverse. | |
| Altă toxicitate hematologică de grad 4§ sau toxicitate de grad 3 care nu poate fi tratată | Se va întrerupe administrarea bendamustinei. Odată ce toxicitatea s-a remis la grad ≤ 2 sau la valorile inițiale, se va relua administrarea bendamustinei în doze de 70 mg/m2. Se va întrerupe definitiv administrarea bendamustinei dacă sunt necesare reduceri suplimentare ale dozelor. | Se va întrerupe administrarea Calquence. Odată ce toxicitatea s-a remis la grad ≤ 2 sau la valorile inițiale, se poate relua administrarea Calquence cu doza de inițiere (la prima apariție a reacției adverse) sau cu frecvență redusă în doză de 100 mg zilnic (la a doua sau a treia apariție a reacției adverse).¶ Se va întrerupe definitiv administrarea Calquence la a patra apariție a reacției adverse. |
| Toxicitate non- hematologică de grad 3 sau mai mare | Se va întrerupe administrarea bendamustinei. Odată ce toxicitatea s-a remis la grad 1 sau la valorile inițiale, se va relua administrarea bendamustinei în doze de 70 mg/m2. Se va întrerupe definitiv administrarea bendamustinei dacă sunt necesare reduceri suplimentare ale dozelor. | Se va întrerupe administrarea Calquence. Odată ce toxicitatea s-a remis la grad 2 sau la valorile inițiale, se va relua administrarea Calquence în doza de inițiere (la prima apariție a reacției adverse) sau cu frecvență redusă în doză de 100 mg zilnic (la a doua sau a treia apariție a reacției adverse).¶ Se va întrerupe definitiv administrarea Calquence la a treia apariție a reacției adverse. |
*Reacțiile adverse au fost clasificate pe grade de severitate conform Criteriilor de terminologie comună pentru evenimentele adverse ale Institutului Național Oncologic (National Cancer Institute Common Terminology Criteria for Adverse Events, NCI CTCAE) versiunea 4.03.
†În cazul în care nu au fost prezentate anumite toxicități în acest tabel, vă rugăm să consultați rezumatul caracteristicilor produsului bendamustină.
‡Se va lua în considerare administrarea factorilor de creștere mieloizi înainte de ajustarea dozelor de bendamustină. §Limfopenia de grad 4 este o reacție adversă așteptată în urma administrării tratamentului cu bendamustină și rituximab. Pot fi efectuate modificări ale dozelor doar dacă medicul investigator consideră aceste modificări ca fiind importante clinic, precum în cazul apariției infecțiilor recurente.
¶Dozele pot fi crescute la valorile inițiale conform deciziei medicului investigator dacă pacientul tolerează doze reduse pentru ≥ 4 săptămâni.
Pentru informații suplimentare privind tratamentul toxicităților vă rugăm să consultați rezumatul caracteristicilor produsului fiecărui medicament administrat în asociere cu Calquence.
Interacţiuni
În tabelul 3 sunt prezentate recomandările cu privire la utilizarea Calquence concomitent cu inhibitori sau inductori ai CYP3A (vezi pct. 4.5).
Tabelul 3. Utilizarea concomitent cu inhibitori sau inductori ai CYP3A
| Medicamentul administrat concomitent | Recomandări privind utilizarea Calquence | |
| Inhibitori CYP3A | Inhibitor puternic al CYP3A | Utilizarea concomitentă trebuie evitată. Dacă aceşti inhibitori se vor utiliza pe termen scurt (cum ar fi anti-infecţioasele, utilizate până la şapte zile), tratamentul cu Calquence trebuie întrerupt. |
Inhibitor moderat al CYP3A | Nu este necesară ajustarea dozelor. Pacienţii trebuie monitorizaţi cu atenţie pentru apariţia reacţiilor adverse în cazul administrării de inhibitori moderaţi ai CYP3A. | |
Inhibitor uşor al CYP3A | Nu este necesară ajustarea dozelor. | |
| Inductori ai CYP3A | Inductor puternic al CYP3A | Utilizarea concomitentă trebuie evitată. |
Acalabrutinib sub formă de comprimate se poate administra concomitent cu medicamente care reduc secreția de acid gastric (inhibitori de pompă de protoni, antagoniști ai receptorilor histaminergici H2, medicamente antiacide), spre deosebire de acalabrutinib sub formă de capsule, care prezintă o absorbție redusă atunci când sunt administrate concomitent cu medicamente care reduc secreția de acid gastric (vezi pct. 4.5).
Doze omise
Dacă pacientul omite să-și administreze o doză de Calquence şi au trecut mai mult de 3 ore de la momentul programat pentru administrare, pacientul trebuie instruit să ia următoarea doză conform schemei terapeutice obişnuite. Nu trebuie administrată o doză dublă de Calquence pentru a compensa doza omisă.
Grupe speciale de pacienți
Vârstnici
Nu este necesară ajustarea dozelor la pacienţi vârstnici (vârsta ≥ 65 ani) (vezi pct. 5.2).
Insuficienţă renală
Nu s-au efectuat studii clinice specifice la pacienţi cu insuficiență renală. Studiile clinice cu Calquence au inclus pacienţi cu insuficiență renală uşoară sau moderată. Nu este necesară ajustarea dozei la pacienţii cu insuficienţă renală uşoară sau moderată (clearance al creatininei mai mare de 30 ml/min). Trebuie menţinută o hidratare adecvată şi nivelurile serice ale creatininei trebuie monitorizate periodic. La pacienţii cu insuficiență renală severă (clearance al creatininei < 30 ml/min) se va administra Calquence numai dacă beneficiile tratamentului depăşesc riscurile şi aceşti pacienţi trebuie monitorizați cu atenţie pentru apariția semnelor de toxicitate. Nu există date provenite de la pacienţii cu insuficienţă renală severă sau de la pacienţii dializaţi (vezi pct. 5.2).
Insuficiență hepatică
Nu există recomandări privind ajustarea dozelor la pacienţii cu insuficiență hepatică uşoară sau moderată (Clasificarea Child-Pugh clasa A, Clasificarea Child-Pugh clasa B sau valori ale bilirubinei totale de 1,5 până la 3 ori mai mari decât limita superioară a valorilor normale [LSVN] şi orice valori ale AST). Cu toate acestea, pacienţii cu insuficiență hepatică moderată trebuie atent monitorizaţi pentru semne de toxicitate. Nu se recomandă utilizarea Calquence la pacienţi cu insuficiență hepatică severă (Clasificarea Child-Pugh clasa C sau valori ale bilirubinei totale de >3 ori mai mari decât LSVN şi orice valori ale AST) (vezi pct. 5.2).
Boală cardiacă severă
Pacienţii cu boli cardiovasculare severe au fost excluşi din studiile clinice privind Calquence.
Copii și adolescenți
Siguranţa şi eficacitatea administrării Calquence la copii şi adolescenţi cu vârsta cuprinsă între 0 şi 18 ani nu au fost încă stabilite. Nu sunt disponibile date.
Mod de administrare
Calquence este indicat pentru administrare pe cale orală. Comprimatele trebuie înghiţite întregi, cu apă, la aproximativ acelaşi moment în fiecare zi, împreună cu sau fără alimente (vezi pct. 4.5). Comprimatele nu trebuie mestecate, sfărâmate, dizolvate sau divizate.
4.3 Contraindicaţii
Hipersensibilitate la substanţa activă sau la oricare dintre excipienţii enumeraţi la punctul 6.1.
4.4 Atenţionări şi precauţii speciale pentru utilizare
Hemoragii
La pacienţii cu malignităţi hematologice trataţi cu Calquence în monoterapie sau în asociere cu alte medicamente au apărut evenimente hemoragice majore, inclusiv hemoragii gastro-intestinale sau la nivelul sistemului nervos central, unele dintre acestea având consecinţe letale. Aceste evenimente au survenit la pacienţi cu, dar şi fără trombocitopenie. Per ansamblu, evenimentele de sângerare au fost de tip mai puţin sever, cum ar fi apariţia echimozelor şi peteşiilor (vezi pct. 4.8).
Mecanismul de la baza evenimentelor de sângerare nu este bine înţeles.
Pacienţii trataţi cu agenţi antitrombotici pot fi expuşi unui risc crescut de apariţie a hemoragiilor. Dacă este necesar din punct de vedere medical, se recomandă prudență în administrare concomitent cu medicamente antitrombotice şi trebuie avută în vedere monitorizarea suplimentară a pacienţilor pentru apariția eventualelor semne de sângerare. Warfarina sau alți inhibitori ai vitaminei K nu trebuie administrați concomitent cu Calquence.
În cazul unei intervenţii chirurgicale, trebuie analizate beneficiile şi riscurile întreruperii tratamentului cu Calquence timp de cel puţin 3 zile înainte şi după intervenţie.
Infecții
La pacienţii cu malignităţi hematologice trataţi cu Calquence în monoterapie sau în asociere cu alte medicamente au apărut cazuri de infecţii grave (bacteriene, virale sau fungice), inclusiv evenimente letale. Aceste infecții au apărut preponderent în absența neutropeniei, infecția neutropenică fiind raportată la 10,1% dintre pacienții care au primit tratament în monoterapie și la 26,8% dintre pacienții care au primit tratamentul asociat. Au fost înregistrate cazuri de infecţii determinate de reactivarea virusului hepatitic B (VHB) şi a virusului herpes zoster (VHZ), cazuri de aspergiloză şi leucoencefalopatie multifocală progresivă (LMP) (vezi pct. 4.8).
Reactivare virală
S-au raportat cazuri de reactivare virală a hepatitei B la pacienţii cărora li se administra Calquence. Înainte de iniţierea tratamentului cu Calquence trebuie determinat statusul serologic al virusului hepatitic B (VHB). Dacă pacienţii sunt pozitivi la testul serologic pentru hepatita B, înainte de a începe tratamentul trebuie consultat un medic specialist în hepatologie şi pacientul trebuie monitorizat şi gestionat conform standardelor medicale locale pentru a se preveni reactivarea hepatitei B.
Au fost raportate cazuri de apariţie a leucoencefalopatiei multifocale progresive (LMP), inclusiv cazuri letale, după administrarea Calquence în contextul utilizării anterioare sau concomitente a unei terapii imunosupresoare. Medicii trebuie să ia în considerare LMP în diagnosticul diferenţial al pacienţilor cu semne sau simptome noi sau agravate de natură neurologică, cognitivă sau comportamentală. Dacă se suspicionează LMP, trebuie efectuate evaluările diagnostice corespunzătoare şi tratamentul cu Calquence trebuie întrerupt până la excluderea LMP. Dacă există orice suspiciune, se recomandă trimiterea pacientului la neurolog în vederea efectuării de teste diagnostice adecvate pentru LMP, inclusiv RMN, de preferinţă cu substanță de contrast, testarea lichidului cefalorahidian (LCR) pentru prezenţa ADN-ului viral al JC şi trebuie avută în vedere repetarea evaluărilor neurologice.
În cazul pacienţilor cu risc crescut de infecţii oportuniste trebuie avut în vedere tratamentul profilactic în conformitate cu standardul de îngrijire. Pacienţii trebuie monitorizaţi pentru apariţia semnelor şi simptomelor de infecţie şi trataţi după cum este adecvat din punct de vedere medical.
Citopenii
La pacienţii cu malignităţi hematologice trataţi cu Calquence în monoterapie sau în asociere cu alte medicamente au fost înregistrate citopenii de gradul 3 sau 4 , inclusiv neutropenie, anemie şi trombocitopenie apărute în cursul tratamentului. Valorile hemoleucogramei trebuie monitorizate după cum este indicat din punct de vedere medical (vezi pct. 4.8).
A doua malignitate primară
La pacienţii cu malignităţi hematologice trataţi cu Calquence în monoterapie sau în asociere cu alte medicamente a apărut şi a doua malignitate primară, inclusiv cancere cutanate sau non-cutanate. Cazurile de cancer cutanat au fost frecvent raportate. Pacienţii trebuie monitorizaţi pentru apariţia cancerelor cutanate şi sfătuiţi să se protejeze de expunerea la soare (vezi pct. 4.8).
Fibrilaţie atrială
În rândul pacienţilor cu malignităţi hematologice trataţi cu Calquence în monoterapie sau în asociere cu alte medicamente au apărut cazuri de fibrilaţie atrială/flutter atrial. Pacienţii trebuie monitorizaţi pentru apariţia simptomelor (de exemplu, palpitaţii, ameţeală, sincopă, durere toracică, dispnee) de fibrilaţie atrială şi flutter atrial, cu efectuarea unei ECG atunci când este indicat medical (vezi pct. 4.5 şi 4.2). La pacienţii care dezvoltă fibrilaţie atrială în timpul tratamentului cu Calquence, trebuie luată în considerare o evaluare detaliată a riscului de afecţiuni tromboembolice. La pacienţii cu risc înalt de afecţiuni tromboembolice, trebuie avut în vedere tratamentul strict controlat cu anticoagulante şi trebuie luate în considerare alte opţiuni terapeutice decât Calquence.
Sindrom de liză tumorală
Sindromul de liză tumorală (SLT) a fost raportat în cazul administrării Calquence. Pacienții la risc pentru SLT (de exemplu, prezența masei tumorale mari la momentul inițial) trebuie evaluați pentru un potențial risc de SLT și trebuie monitorizați atent conform indicațiilor clinice.
Boală pulmonară interstițială/pneumonită
Boala interstițială pulmonară (BIP)/pneumonita a fost raportată la pacienții cărora li s-a administrat tratament cu Calquence în asociere cu bendamustină și rituximab pentru LCM. Pacienții trebuie monitorizați pentru apariția simptomelor care indică prezența BIP/pneumonită (de exemplu, tuse, dispnee sau hipoxie) și în cazul manifestării BIP/pneumonitei, pacienții trebuie tratați conform indicațiilor clinice.
Alte medicamente
Administrarea de inhibitori puternici ai CYP3A concomitent cu Calquence poate determina creşterea expunerii la acalabrutinib şi, în consecinţă, creşterea riscului de apariţie a toxicităţilor. Invers, administrarea de inductori ai CYP3A concomitent cu Calquence poate determina scăderea expunerii la acalabrutinib, cu riscul implicit de pierdere a eficacităţii. Administrarea concomitentă cu inhibitori puternici ai CYP3A trebuie evitată. Dacă acești inhibitori vor fi utilizați pe termen scurt (cum sunt medicamentele anti-infecțioase administrate până la 7 zile), tratamentul cu Calquence trebuie întrerupt. În cazul în care este administrat un inhibitor moderat al CYP3A, pacienţii trebuie monitorizaţi îndeaproape pentru apariţia semnelor de toxicitate (vezi pct. 4.2 şi 4.5). Utilizarea concomitentă cu inductori puternici ai CYP3A4 trebuie evitată datorită riscului de ineficacitate a tratamentului.
Calquence conţine sodiu
Acest medicament conţine sodiu mai puţin de 1 mmol (23 mg) per doză, adică practic „nu conţine sodiu”.
4.5 Interacţiuni cu alte medicamente şi alte forme de interacţiune
Acalabrutinib şi metabolitul său activ sunt metabolizaţi preponderent de enzima 3A4 a citocromului P450 (CYP3A4) şi ambii constituie substraturi pentru gp-P şi proteina de rezistență în cancerul mamar (breast cancer resistance protein, BCRP).
Substanţe active care pot creşte concentraţiile plasmatice de acalabrutinib
Inhibitori ai CYP3A/gp-P
Administrarea concomitentă cu un inhibitor puternic al CYP3A/gp-P (itraconazol în doză de 200 mg o dată pe zi, timp de 5 zile) a crescut valorile Cmax şi ASC pentru acalabrutinib de 3,9 ori şi, respectiv, de 5,0 ori la voluntari sănătoşi (N=17).
Administrarea concomitentă cu inhibitori puternici ai CYP3A/gp-P trebuie evitată. În cazul în care este necesară utilizarea pe termen scurt a inhibitorilor puternici de CYP3A/gp-P (de exemplu, ketaconazol, conivaptan, claritromicină, indinavir, itraconazol, ritonavir, telaprevir, posaconazol, voriconazol), tratamentul cu Calquence trebuie întrerupt (vezi pct. 4.2).
Administrarea concomitentă cu inhibitori moderați ai CYP3A (fluconazol 400 mg în doză unică sau isavuconazol 200 mg în doză repetată timp de 5 zile) la voluntari sănătoși a crescut valorile Cmax și ASC pentru acalabrutinib de 1,4 până la 2 ori, în timp ce valorile Cmax și ASC pentru metabolitul activ ACP-5862 au scăzut de 0,65 ori până la 0,88 ori, comparativ cu administrarea acalabrutinib în monoterapie. Nu este necesară ajustarea dozei în cazul administrării concomitente cu inhibitori moderați ai CYP3A. Pacienții trebuie monitorizați îndeaproape pentru apariția reacțiilor adverse (vezi pct. 4.2).
Substanţe active care pot scădea concentraţiile plasmatice de acalabrutinib
Inductori ai CYP3A
Administrarea concomitentă cu un inductor puternic al CYP3A (rifampicină în doză de 600 mg o dată pe zi, timp de 9 zile) a scăzut valorile Cmax şi ASC pentru acalabrutinib cu 68% şi, respectiv, cu 77% la voluntari sănătoşi (N=24).
Administrarea concomitentă cu inductori potenţi ai activităţii CYP3A (de exemplu, fenitoină, rifampicină, carbamazepină) trebuie evitată. Utilizarea concomitentă a sunătorii trebuie evitată deoarece poate scădea în mod impredictibil concentraţiile plasmatice de acalabrutinib.
Medicamente care scad secreţia de acid gastric
Nu au fost observate diferențe relevante clinic în ceea ce privește farmacocinetica acalabrutinib atunci când acesta a fost administrat sub formă de comprimate 100 mg concomitent cu un inhibitor al pompei de protoni (rabeprazol 20 mg de două ori pe zi, timp de 3 zile). Acalabrutinib sub formă de comprimate poate fi administrat concomitent cu agenți care reduc secreția de acid gastric (inhibitori de pompă de protoni, antagoniști ai receptorilor histaminergici H2, medicamente antiacide), spre deosebire de acalabrutinib sub formă de capsule, care prezintă o absorbție redusă atunci când sunt administrate concomitent cu medicamente care reduc secreția de acid gastric.
Substanţe active ale căror concentraţii plasmatice pot fi modificate de Calquence
Substraturi pentru CYP3A
Pe baza datelor din studiile in vitro, nu se poate exclude posibilitatea ca acalabrutinib să inhibe CYP3A4 la nivel intestinal şi să crească astfel expunerea la medicamentele-substrat pentru CYP3A4 susceptibile a fi metabolizate de enzimele CYP3A din intestin. Se recomandă prudenţă în cazul utilizării acalabrutinib concomitent cu medicamente substrat pentru CYP3A4, cu interval terapeutic îngust şi care se administrează pe cale orală (precum ciclosporină, ergotamină, pimozidă).
Efectul acalabrutinib asupra medicamentelor substrat pentru CYP1A2
Studiile in vitro indică faptul că acalabrutinib induce activitatea CYP1A2. Administrarea acalabrutinib concomitent cu substraturi pentru CYP1A2 (de exemplu, teofilină, cafeină) poate scădea expunerea la aceste medicamente.
Efectele acalabrutinib şi ale metabolitului său activ, ACP-5862, asupra sistemelor transportoare de medicamente
Acalabrutinib poate creşte expunerea la medicamentele substrat pentru BCRP (de exemplu, metotrexatul) administrate simultan, prin inhibiţia BCRP de la nivel intestinal (vezi pct. 5.2). Pentru a reduce la minimum posibilitatea unei interacţiuni la nivel gastrointestinal (GI), medicamentele substrat pentru BCRP cu administrare pe cale orală şi cu interval terapeutic îngust, precum metotrexatul, trebuie administrate cu cel puţin 6 ore înainte sau după acalabrutinib.
ACP-5862 poate creşte expunerea la medicamentele substrat pentru MATE1 (de exemplu, metforminul) administrate simultan, prin inhibiţia MATE1 (vezi pct. 5.2). Pacienţii trataţi concomitent cu medicamente a căror eliminare depinde de transportorul MATE1 (precum metforminul) trebuie monitorizaţi pentru depistarea semnelor de modificare a tolerabilităţii ca urmare a creşterii expunerii la medicaţia administrată concomitent cu Calquence.
4.6 Fertilitatea, sarcina şi alăptarea
Femeile aflate la vârsta fertilă
Femeile cu potenţial fertil trebuie sfătuite să nu rămână însărcinate pe durata tratamentului cu Calquence.
Sarcina
Nu există date sau există un volum limitat de date privind utilizarea acalabrutinib la femeile gravide. Pe baza rezultatelor din studiile la animale, este posibil să existe un risc pentru făt asociat cu expunerea la acalabrutinib pe durata sarcinii. Au fost observate cazuri de distocie (travaliu dificil sau prelungit) la şobolan, iar administrarea la femele de iepure gestante s-a asociat cu reducerea creşterii fetale (vezi pct. 5.3).
Calquence nu trebuie utilizat pe durata sarcinii decât dacă starea clinică a femeii impune tratamentul cu acalabrutininb.
Alăptarea
Nu se cunoaşte dacă acalabrutinib se excretă în laptele uman. Nu există date cu privire la efectele acalabrutinib asupra sugarului sau asupra producţiei de lapte. Prezenţa acalabrutinib şi a metabolitului său activ a fost depistată în laptele matern al femelelor de şobolan. Nu se poate exclude un risc pentru sugari. Mamele aflate în perioada de alăptare sunt sfătuite să nu alăpteze pe durata tratamentului cu Calquence şi timp de încă 2 zile după administrarea ultimei doze.
Fertilitatea
Nu există date privind efectele Calquence asupra fertilităţii la om. În cadrul unui studiu non-clinic în care s-a administrat acalabrutinib la şobolani masculi şi femele nu au fost observate efecte adverse asupra parametrilor de fertilitate (vezi pct. 5.3).
4.7 Efecte asupra capacităţii de a conduce vehicule şi de a folosi utilaje
Calquence nu are nicio influenţă sau are influenţă neglijabilă asupra capacităţii de a conduce vehicule sau de a folosi utilaje. Cu toate acestea, pe durata tratamentului cu acalabrutinib au fost raportate stări de oboseală şi ameţeli, iar pacienţii care manifestă aceste simptome trebuie sfătuiţi să nu conducă vehicule sau să folosească utilaje decât după dispariţia acestora.
4.8 Reacţii adverse
Rezumatul profilului de siguranţă
Calquence în monoterapie
În rândul celor 1478 pacienţi trataţi cu Calquence în monoterapie, cele mai frecvente (≥ 20%) reacţii adverse induse de medicament (RAIM) de orice grad au fost infecţiile, diareea, cefaleea, durerea musculoscheletică, echimozele, tusea, artralgia, fatigabilitatea, greața şi erupţiile cutanate tranzitorii. Cel mai frecvent raportate (≥ 5%) reacţii adverse induse de medicament cu severitate de grad ≥ 3 au fost infecţiile, leucopenia, neutropenia, anemia, a doua malignitate primară şi trombocitopenia.
Calquence în asociere cu obinutuzmab
În rândul celor 223 de pacienţi trataţi cu Calquence în asociere cu obinutuzumb, cele mai frecvente (≥ 20%) RAIM de orice grad au fost infecţiile, durerea musculo-scheletică, diareea, cefaleea, leucopenia, neutropenia, tusea, fatigabilitatea, artralgia, greaţa, ameţeala şi constipația. Cel mai frecvent raportate (≥ 5%) reacţii adverse induse de medicament cu severitate de grad ≥ 3 au fost leucopenia, neutropenia, infecțiile, trombocitopenia şi anemia.
Calquence în asociere cu venetoclax
În rândul celor 291 de pacienți tratați cu Calquence în asociere cu venetoclax, cele mai frecvente (≥ 20%) RAIM de orice grad au fost infecțiile, neutropenia, cefaleea, echimozele, diareea și durerea musculoscheletică. Cel mai frecvent raportată (≥ 5%) RAIM cu severitate de grad ≥ 3 a fost neutropenia.
Calquence în asociere cu venetoclax și obinutuzumab
În rândul celor 284 de pacienți tratați cu Calquence în asociere cu venetoclax și obinutuzumab, cele mai frecvente (≥ 20%) RAIM de orice grad au fost infecțiile, neutropenia, cefaleea, echimozele, diareea, greața și durerea musculo-scheletică. Cel mai frecvent raportate (≥ 5%) RAIM cu severitate de grad ≥ 3 au fost neutropenia și trombocitopenia.
Calquence în asociere cu bendamustină și rituximab
În rândul celor 297 de pacienți tratați cu Calquence în asociere cu bendamustină și rituximab, cele mai frecvente (≥ 20%) RAIM de orice grad au fost neutropenia, greața, erupția cutanată tranzitorie, diareea, durerea musculo-scheletică, cefaleea, fatigabilitatea, vărsăturile, constipația, anemia și trombocitopenia.
Cel mai frecvent raportate (≥ 5%) reacții adverse induse de medicament cu severitate de grad ≥ 3 au fost neutropenia, erupția cutanată tranzitorie, trombocitopenia, anemia, pneumonia, a doua malignitate primară, hipertensiunea arterială și a doua malignitate primară cu excepția cancerului cutanat de alt tip decât melanom.
Lista tabelară a reacțiilor adverse
Reacțiile adverse induse de medicament (RAIM) identificate în studiile clinice derulate la pacienți cărora li s-a administrat Calquence în monoterapie sau în terapie asociată pentru tratamentul malignităților hematologice sunt prezentate în tabelele următoare. Durata mediană a tratamentului cu Calquence în monoterapie conform datelor cumulate din studii a fost de 38,2 luni. Durata mediană a tratamentului cu Calquence în asociere cu bendamustină și rituximab a fost de 28,6 luni. Durata mediană a tratamentului cu Calquence la pacienții tratați cu Calquence în asociere cu venetoclax cu sau fără obinutuzumab a fost de 12,9 luni.
Reacţiile adverse induse de medicament sunt enumerate conform sistemului MedDRA de clasificare pe aparate, sisteme şi organe (ASO). În cadrul fiecărei clase de aparate, sisteme şi organe, reacţiile adverse sunt enumerate în funcţie de frecvenţă, începând cu cele mai frecvente. De asemenea, categoriile corespunzătoare de frecvenţă pentru fiecare RAIM sunt definite astfel: foarte frecvente (≥ 1/10); frecvente (≥ 1/100 şi < 1/10); mai puţin frecvente (≥ 1/1000 şi < 1/100); rare (≥ 1/10 000 şi < 1/1000); foarte rare (< 1/10 000); cu frecvenţă necunoscută (care nu poate fi estimată din datele disponibile). În cadrul fiecărei categorii de frecvenţă, reacţiile adverse sunt prezentate în ordinea descrescătoare a gravităţii.
Tabelul 4. Reacții adverse induse de medicament* la pacienţi cu malignităţi hematologice trataţi cu acalabrutinib în monoterapie (N=1478)
| ASO MedDRA | Termen MedDRA | Toate gradele (%) | Grad CTCAE ≥ 3* (%) |
|---|---|---|---|
| Infecții și infestări | Infecții la nivelul tractului respirator superior | Foarte frecvente (25,8) | 1,2 |
| Pneumonie | Foarte frecvente (15,8) | 8,7 | |
| Sinuzită | Foarte frecvente (11,4) | 0,4 | |
| Infecţie la nivelul tractului urinar | Frecvente (9,9) | 1,8 | |
| Bronșită | Frecvente (9,7) | 0,6 | |
| Infecții cu virus herpetic† | Frecvente (9,1) | 0,9 | |
| Rinofaringită | Frecvente (8,3) | 0 | |
| Infecţii cu Aspergillus† | Mai puţin frecvente (0,7) | 0,6 | |
| Reactivarea hepatitei B | Mai puţin frecvente (0,4) | 0,3 | |
| Tumori benigne, maligne şi de tip nedeterminat | A doua malignitate primară (ADMP)† Cancer cutanat de alt tip decât melanomul† ADMP cu excepţia cancerului cutanat de alt tip decât melanomul† | Foarte frecvente (17,6) Frecvente (9,9) Frecvente (9,7) | 6,7 1,4 5,5 |
| Tulburări hematologice şi limfatice | Neutropenie† | Foarte frecvente (19,4) | 17,5 |
| Anemie† | Foarte frecvente (17,1) | 9,5 | |
| Trombocitopenie† | Foarte frecvente (11,5) | 6,2 | |
| Limfocitoză | Mai puţin frecvente (0,5) | 0,3 | |
| Tulburări metabolice și de nutriție | Sindrom de liză tumorală | Mai puţin frecvente (0,5) | 0,4 |
| Tulburări ale sistemului nervos | Cefalee | Foarte frecvente (36,5) | 1,2 |
| Amețeală | Foarte frecvente (13,9) | 0,1 | |
| Tulburări cardiace | Fibrilaţie atrială/flutter atrial† † | Frecvente (7,4) | 2,3 |
| Tulburări vasculare | Echimoze† Contuzii Peteşii Echimoze | Foarte frecvente (30,9) Foarte frecvente (20,7) Frecvente (8,9) Frecvente (5,7) | 0 0 0 0 |
| Hemoragie/hematom† Hemoragie gastro-intestinală Hemoragie intracraniană | Foarte frecvente (16,3) Mai puțin frecvente (0,9) Mai puțin frecvente (0,1) | 3,2 0,7 0,1 | |
| Hipertensiune arterialㆠ| Foarte frecvente (11,9) | 4,9 | |
| Epistaxis | Frecvente (8,0) | 0,3 | |
| Tulburări gastro- intestinale | Diaree | Foarte frecvente (36,7) | 2,6 |
| Greaţă | Foarte frecvente (21,8) | 0,8 | |
| Constipaţie | Foarte frecvente (15,2) | 0,1 | |
| Durere abdominalㆠ| Foarte frecvente (14,5) | 1,2 | |
| Vărsături | Foarte frecvente (14,0) | 0,7 | |
| Afecțiuni cutanate și ale țesutului subcutanat | Erupţie cutanată tranzitorie† | Foarte frecvente (20,3) | 0,9 |
| Tulburări musculo-scheletice şi ale ţesutului conjunctiv | Durere musculo-scheleticㆠ| Foarte frecvente (31,9) | 1,8 |
| Artralgie | Foarte frecvente (24,0) | 0,9 | |
| Tulburări generale şi la nivelul locului de administrare | Fatigabilitate | Foarte frecvente (23,6) | 2,0 |
| Astenie | Frecvente (7,0) | 0,9 | |
| Investigaţii diagnostice§ (constatări bazate pe rezultatele testelor) | Scăderea valorilor hemoglobinei± | Foarte frecvente (47,4) | 10,8 |
| Scăderea numărului absolut de neutrofile (NAN)± | Foarte frecvente (43,9) | 24,0 | |
| Scăderea numărului de trombocite± | Foarte frecvente (36,9) | 9,5 |
*Conform Criteriilor de terminologie comună pentru evenimentele adverse ale Institutului Național Oncologic (National Cancer Institute Common Terminology Criteria for Adverse Events, NCI CTCAE), versiunea 4.03. †Include mai mulți termeni pentru RAIM.
±Reprezintă incidența valorilor rezultatelor de laborator, nu a evenimentelor adverse raportate.
§Prezentate pe grade de severitate CTCAE.
Tabelul 5. Reacţii adverse induse de medicament* la pacienţii cu malignităţi hematologice trataţi cu acalabrutinib ca terapie asociată (N=1095)
| Calquence + obinutuzumab N=223 | Calquence + BR N=297 | Calquence + venetoclax N=291 | Calquence + venetoclax + obinutuzumab N=284 | |||||
| ASO MedDRA și Termen MedDRA | Toate gradele (%) | Grad ≥ 3* (%) | Toate gradele (%) | Grad ≥ 3* (%) | Toate gradele (%) | Grad ≥ 3* (%) | Toate gradele (%) | Grad ≥ 3* (%) |
| Infecții și infestări | ||||||||
|---|---|---|---|---|---|---|---|---|
| Infecții ale tractului respirator superior | Foarte frecvente (31,4) | 1,8 | Foarte frecvente (18,2) | 0,3 | Frecvente (8,2) | 0,3 | Frecvente (6,3) | 0 |
| Sinuzită | Foarte frecvente (15,2) | 0,4 | Frecvente (6,4) | 0 | Frecvente (2,7) | 0 | Frecvente (2,5) | 0 |
| Rinofaringită | Foarte frecvente (13,5) | 0,4 | Frecvente (5,4) | 0 | Frecvente (1,4) | 0 | Frecvente (1,1) | 0 |
| Infecții ale tractului urinar | Foarte frecvente (13) | 0,9 | Foarte frecvente (11,1) | 1,7 | Frecvente (3,1) | 0 | Frecvente (6,0) | 0,4 |
| Pneumonie | Foarte frecvente (10,8) | 5,4 | Foarte frecvente (16,2) | 8,8 | Frecvente (3,8) | 1,4 | Frecvente (5,3) | 3,9 |
| Bronșită | Frecvente (9,9) | 0 | Frecvente (6,4) | 0,3 | Frecvente (2,1) | 0 | Frecvente (2,5) | 0 |
| Infecții cu virus herpetic† | Frecvente (6,7) | 1,3 | Foarte frecvente (12,8) | 1,0 | Frecvente (4,8) | 0 | Frecvente (3,5) | 0,4 |
| Leucoencefalopatie multifocală progresivă | Mai puțin frecvente (0,4) | 0,4 | Cu frecvență necunoscută | 0 | Cu frecvență necunoscută | 0 | Cu frecvență necunoscută | 0 |
| Reactivarea hepatitei B | Mai puțin frecvente (0,9) | 0,1 | Frecvente (1,3) | 0,3 | Cu frecvență necunoscută | 0 | Cu frecvență necunoscută | 0 |
Infecții cu Aspergillus† | Cu frecvență necunoscută | 0 | Mai puțin frecvente (0,3) | 0,3 | Cu frecvență necunoscută | 0 | Mai puțin frecvente (0,4) | 0,4 |
| Tumori benigne, maligne și de tip nedeterminat | ||||||||
A doua malignitate primară†(ADMP) Cancer cutanat de alt tip decât melanom† ADMP cu excepția cancerului cutanat de alt tip decât melanom† | Foarte frecvente (13) Frecvente (7,6) Frecvente (6,3) | 4,0 0,4 3,6 | Foarte frecvente (17,8) Foarte frecvente (11,1) Frecvente (9,8) | 7,4 2,0 5,4 | Frecvente (5,2) Frecvente (3,1) Frecvente (2,7) | 1,7 0 1,7 | Frecvente (4,2) Frecvente (1,8) Frecvente (2,5) | 1,8 0,4 1,4 |
| Tulburări hematologice și limfatice | ||||||||
| Neutropenie† | Foarte frecvente (31,8) | 30 | Foarte frecvente (54,9) | 50,2 | Foarte frecvente (37,1) | 32,3 | Foarte frecvente (50,4) | 46,1 |
| Trombocitopenie† | Foarte frecvente (13,9) | 9 | Foarte frecvente (22,9) | 9,8 | Frecvente (5,8) | 2,1 | Foarte frecvente (12,3) | 9,2 |
| Anemie† | Foarte frecvente (11,7) | 5,8 | Foarte frecvente (24,2) | 9,4 | Frecvente (6,9) | 3,8 | Frecvente (4,6) | 2,1 |
| Limfocitoză | Mai puțin frecvente (0,4) | 0,4 | Mai puțin frecvente (0,7) | 0 | Cu frecvență necunoscută | 0 | Mai puțin frecvente (0,7) | 0,4 |
| Tulburări metabolice și de nutriție | ||||||||
| Sindrom de liză tumorală | Frecvente (1,8) | 1,3 | Frecvente (1,3) | 1,3 | Mai puțin frecvente (0,3) | 0,3 | Mai puțin frecvente (0,4) | 0,4 |
| Tulburări ale sistemului nervos | ||||||||
| Cefalee | Foarte frecvente (43) | 0,9 | Foarte frecvente (30,3) | 1,3 | Foarte frecvente (35,1) | 1,4 | Foarte frecvente (28,2) | 0,4 |
| Amețeală | Foarte frecvente (23,8) | 0 | Foarte frecvente (14,5) | 0,7 | Frecvente (5,5) | 0 | Frecvente (6,7) | 0 |
| Tulburări cardiace | ||||||||
| Fibrilație atrială/flutter atrial† | Frecvente (3,1) | 0,9 | Frecvente (6,7) | 4,0 | Mai puțin frecvente (0,7) | 0,3 | Frecvente (2,1) | 0,7 |
| Tulburări vasculare | ||||||||
Echimoze† Contuzii Peteșii Echimoze | Foarte frecvente (38,6) Foarte frecvente (27,4) Foarte frecvente (11,2) Frecvente (3,1) | 0 0 0 0 | Foarte frecvente (14,1) Foarte frecvente (11,1) Frecvente (2,0) Frecvente (3,0) | 0,3 0 0 0,3 | Foarte frecvente (20,6) Foarte frecvente (14,1) Frecvente (4,8) Frecvente (2,7) | 0 0 0 0 | Foarte frecvente (21,8) Foarte frecvente (16,2) Frecvente (5,3) Frecvente (3,9) | 0 0 0 0 |
Hemoragie/hematom Hemoragie gastro- intestinală Hemoragie intracraniană | Foarte frecvente (17,5) Frecvente (3,6) Mai puțin frecvente (0,9) | 1,3 0,9 0 | Foarte frecvente (15,5) Mai puțin frecvente (0,3) Cu frecvență necunoscută | 1,0 0 0 | Frecvente (8,9) Mai puțin frecvente (0,7) Cu frecvență necunoscută | 0,7 0,3 0 | Frecvente (8,5) Cu frecvență necunoscută Cu frecvență necunoscută | 1,1 0 0 |
| Hipertensiune arterialㆠ| Foarte frecvente (13,5) | 3,6 | Foarte frecvente (12,5) | 5,7 | Frecvente (4,1) | 2,7 | Frecvente (3,9) | 2,1 |
| Epistaxis | Frecvente (8,5) | 0 | Frecvente (2,7) | 0 | Frecvente (1,7) | 0 | Frecvente (4,2) | 0 |
| Tulburări respiratorii, toracice și mediastinale | ||||||||
| Pneumonită± | - | - | Frecvente (2,4) | 0,3 | - | - | - | - |
| Tulburări gastro-intestinale | ||||||||
| Diaree | Foarte frecvente (43,9) | 4.5 | Foarte frecvente (37,4) | 3,0 | Foarte frecvente (32,6) | 1,7 | Foarte frecvente (36,3) | 1,4 |
| Greață | Foarte frecvente (26,9) | 0 | Foarte frecvente (42,8) | 1,3 | Foarte frecvente (14,8) | 0 | Foarte frecvente (21,8) | 0,7 |
| Constipație | Foarte frecvente (20,2) | 0 | Foarte frecvente (24,6) | 1,0 | Frecvente (6,5) | 0,3 | Frecvente (8,1) | 0 |
| Vărsături | Foarte frecvente (19,3) | 0,9 | Foarte frecvente (25,6) | 0,7 | Frecvente (5,5) | 0 | Frecvente (6,7) | 0 |
| Durere abdominalㆠ| Foarte frecvente (14,8) | 1,3 | Foarte frecvente (12,1) | 2,0 | Frecvente (7,9) | 1,0 | Frecvente (8,1) | 0,7 |
| Tulburări cutanate și ale țesutului subcutanat | ||||||||
| Erupție cutanată tranzitorie† | Foarte frecvente (30,9) | 1,8 | Foarte frecvente (39,1) | 9,8 | Foarte frecvente (12,0) | 0,3 | Foarte frecvente (16,2) | 1,1 |
| Tulburări musculo-scheletice și ale țesutului conjunctiv | ||||||||
| Durere musculo- scheleticㆠ| Foarte frecvente (44,8) | 2,2 | Foarte frecvente (34,3) | 3,7 | Foarte frecvente (24,1) | 0,7 | Foarte frecvente (21,8) | 1,1 |
| Artralgie | Foarte frecvente (26,9) | 1,3 | Foarte frecvente (17,5) | 0,7 | Foarte frecvente (12,7) | 1,0 | Foarte frecvente (10,9) | 0,4 |
| Tulburări generale și la nivelul locului de administrare | ||||||||
| Fatigabilitate | Foarte frecvente (30,5) | 1,8 | Foarte frecvente (29,3) | 2,7 | Foarte frecvente (14,8) | 0,3 | Foarte frecvente (14,4) | 0 |
| Astenie | Frecvente (7,6) | 0,4 | Foarte frecvente (10,4) | 1,0 | Frecvente (4,1) | 0 | Frecvente (3,2) | 0 |
| Investigații diagnostice¶ | ||||||||
| Scăderea numărului absolut de neutrofile§ | Foarte frecvente (57,4) | 35 | Foarte frecvente (76,8) | 56,6 | Foarte frecvente (78,0) | 38,1 | Foarte frecvente (81,7) | 53,5 |
| Scăderea numărului de trombocite§ | Foarte frecvente (46,2) | 10,8 | Foarte frecvente (69,4) | 17,8 | Foarte frecvente (42,6) | 5,2 | Foarte frecvente (54,9) | 13,7 |
| Scăderea valorilor hemoglobinei§ | Foarte frecvente (43,9) | 9 | Foarte frecvente (79,5) | 10,8 | Foarte frecvente (34,7) | 6,5 | Foarte frecvente (45,8) | 3,5 |
| Creșterea nivelului alanin aminotransferazei‡ | - | - | Frecvente (9,1) | 4,4 | - | - | - | - |
| Creșterea nivelului aspartat aminotransferazei‡ | - | - | Frecvente (8,1) | 3,0 | - | - | - | - |
*Conform Criteriilor de terminologie comună pentru evenimentele adverse ale Institutului Național Oncologic (National Cancer Institute Common Terminology Criteria for Adverse Events, NCI CTCAE), versiunea 4.03. †Include mai mulți termeni pentru RAIM.
±A fost raportat un eveniment advers care a determinat decesul pacientului.
§Reprezintă incidența valorilor rezultatelor de laborator, nu a evenimentelor adverse raportate.
¶Prezentate pe grade de severitate CTCAE.
‡Reacție adversă raportată doar la grupul de pacienți tratați cu Calquence + BR din studiul ECHO.
Descrierea reacțiilor adverse selectate
Infecții grave raportate la pacienții tratați cu Calquence în asociere cu venetoclax cu sau fără obinutuzumab
În rândul celor 291 de pacienți tratați cu Calquence în asociere cu venetoclax, infecțiile severe (grad ≥ 3) au fost raportate la 12,4% dintre pacienți (cel mai frecvent raportate au fost COVID-19 sau pneumonia asociată COVID-19). Infecțiile care au determinat deces au fost raportate la 3,1% dintre pacienți (cel mai frecvent raportate au fost COVID-19 sau pneumonia asociată COVID-19).
În rândul celor 284 de pacienți tratați cu Calquence în asociere cu venetoclax și obinutuzumab, infecțiile grave (grad ≥ 3) au fost raportate la 23,6% dintre pacienți (cel mai frecvent raportate au fost COVID-19 sau pneumonia asociată COVID-19). Infecțiile care au determinat deces au fost raportate la 5,6% dintre pacienți (cel mai frecvent raportate au fost COVID-19 sau pneumonia asociată COVID-19).
Întreruperea administrării şi reducerea dozei din cauza reacţiilor adverse
În rândul celor 1478 de pacienţi trataţi cu Calquence în monoterapie, cazurile de întrerupere a tratamentului din cauza reacţiilor adverse au fost raportate la 14,6% dintre pacienţi. Principalele reacţii adverse au fost pneumonia, trombocitopenia şi diareea. Reducerile dozei din cauza reacţiilor adverse au fost raportate la 5,9% dintre pacienţi. Principalele reacţii adverse au fost reactivarea hepatitei B, sepsisul şi diareea.
În rândul celor 223 de pacienţi trataţi cu Calquence în asociere cu obinutuzumab, cazurile de întrerupere a tratamentului cu Calquence din cauza reacţiilor adverse au fost raportate la 10,8% dintre pacienţi. Principalele reacţii adverse au fost pneumonia, trombocitopenia şi diareea. Reducerile dozei din cauza reacţiilor adverse au fost raportate la 6,7% dintre pacienţi. Principalele reacţii adverse au fost neutropenia, diareea şi vărsăturile.
În rândul celor 291 de pacienți tratați cu Calquence în asociere cu venetoclax, cazurile de întrerupere definitivă a tratamentului cu Calquence din cauza reacțiilor adverse au fost raportate la 7,6% dintre pacienți și cazurile de reducere a dozelor de Calquence din cauza reacțiilor adverse au fost raportate la 5,8% dintre pacienți. Principalele reacții adverse care au determinat întreruperea definitivă a tratamentului au inclus pneumonia asociată COVID-19 și COVID-19, iar reacția adversă care a determinat reducerea dozelor a fost neutropenia.
În rândul celor 284 de pacienți tratați cu Calquence în asociere cu venetoclax și obinutuzumab, cazurile de întrerupere definitivă a tratamentului cu Calquence din cauza reacțiilor adverse au fost raportate la 13,7% dintre pacienți și cazurile de reducere a dozelor de Calquence din cauza reacțiilor adverse au fost raportate la 6,3% dintre pacienți. Principalele reacții adverse care au determinat întreruperea definitivă a tratamentului au inclus pneumonia asociată COVID-19 și COVID-19, iar reacția adversă care a determinat reducerea dozelor a fost neutropenia.
În rândul celor 297 de pacienți tratați cu Calquence în asociere cu bendamustină și rituximab, cazurile de întrerupere definitivă a tratamentului cu Calquence din cauza reacțiilor adverse au fost raportate la 42,8% dintre pacienți. Principalele reacții adverse au fost COVID-19, pneumonie asociată COVID-19, neutropenie și pneumonie. Reducerile dozei din cauza reacțiilor adverse au fost raportate la 10,1% dintre pacienți. Principalele reacții adverse au inclus neutropenie și greață.
Vârstnici
Dintre cei 1478 de pacienţi din studiile clinice cu Calquence în monoterapie, 42% au avut vârsta peste 65 de ani şi sub 75 de ani şi 20,6% au avut vârsta de 75 de ani sau peste. Nu au fost observate diferenţe relevante clinic între pacienţii cu vârsta ≥ 65 ani şi pacienţii mai tineri în ceea ce priveşte siguranţa sau eficacitatea tratamentului.
Dintre cei 223 de pacienţi din studiile clinice cu Calquence în asociere cu obinutuzumab, 47% au avut vârsta peste 65 de ani şi sub 75 de ani şi 26% au avut vârsta de 75 de ani sau peste. Nu au fost observate diferenţe relevante clinic între pacienţii cu vârsta ≥65 ani şi pacienţii mai tineri în ceea ce priveşte siguranţa sau eficacitatea tratamentului.
Dintre cei 291 de pacienți tratați cu Calquence în asociere cu venetoclax, 28,9% au avut vârsta peste 65 de ani și sub 75 de ani și 4,5% au avut vârsta de 75 de ani sau peste. Nu au fost observate diferențe relevante clinic între pacienții cu vârsta ≥ 65 de ani și pacienții mai tineri, în ceea ce privește siguranța și eficacitatea tratamentului.
Dintre cei 284 de pacienți tratați cu Calquence în asociere cu venetoclax și obinutuzumab, 24% au avut vârsta peste 65 de ani și sub 75 de ani și 6,3% au avut vârsta de 75 de ani sau peste. Nu au fost observate diferențe relevante clinic între pacienții cu vârsta ≥ 65 de ani și pacienții mai tineri, în ceea ce privește siguranța și eficacitatea tratamentului.
Raportarea reacţiilor adverse suspectate
Raportarea reacțiilor adverse suspectate după autorizarea medicamentului este importantă. Acest lucru permite monitorizarea continuă a raportului beneficiu/risc al medicamentului. Profesioniştii din domeniul sănătăţii sunt rugaţi să raporteze orice reacţie adversă suspectată prin intermediul sistemului naţional de raportare, astfel cum este menţionat în Anexa V.
4.9 Supradozaj
Nu există un tratament specific pentru supradozajul cu acalabrutinib şi simptomele supradozajului nu au fost stabilite. În caz de supradozaj, pacienţii trebuie atent monitorizaţi pentru apariţia semnelor sau simptomelor de reacţii adverse şi trebuie iniţiat tratamentul simptomatic corespunzător.
5. PROPRIETĂŢI FARMACOLOGICE
5.1 Proprietăţi farmacodinamice
Grupa farmacoterapeutică: Medicamente antineoplazice, inhibitori de protein-kinază, cod ATC: L01EL02.
Mecanism de acţiune
Acalabrutinib este un inhibitor selectiv al tirozin kinazei Bruton (Bruton tyrosine kinase, TKB). TKB este o moleculă de semnalizare a căilor receptorului pentru antigen al celulelor B (B-cell antigen receptor, BCR) şi ale receptorilor citokinici. Semnalizarea TKB la nivelul celulelor B are ca rezultat supravieţuirea şi proliferarea acestora şi este necesară pentru adeziunea, traficul şi chemotaxia celulelor.
Acalabrutinib şi metabolitul său activ, ACP-5862, formează o legătură covalentă cu un rest de cisteină din situsul activ al TKB, determinând inactivarea ireversibilă a TKB cu interacţiuni minimale în afara țintei (off target).
Efecte farmacodinamice
La pacienţii cu malignităţi de celulă B cărora li s-a administrat acalabrutinib în doză de 100 mg de două ori pe zi, gradul median de ocupare a TKB la starea de echilibru ≥ 95% în sângele periferic a fost menţinut timp de 12 ore, având ca rezultat inactivarea TKB pe parcursul intervalului recomandat între administrările dozelor.
Electrofiziologia cardiacă
Efectul acalabrutinib asupra intervalului QTc a fost evaluat pe 46 de subiecţi sănătoşi de sex masculin şi feminin în cadrul unui studiu clinic, randomizat, dublu-orb, asupra intervalului QT controlat cu placebo şi comparator activ. La administrarea în doză supraterapeutică, de 4 ori mai mare decât doza maximă recomandată, Calquence nu a prelungit intervalul QT/QTc într-o măsură relevantă clinic (de exemplu, ≥ 10 ms) (vezi pct. 4.4, 4.8 şi 5.3).
Eficacitate și siguranță clinică
Pacienţii cu LLC netratată anterior
Calquence în monoterapie sau în asociere cu obinutuzumab
Siguranţa şi eficacitatea Calquence în monoterapie sau în asociere cu obinutuzumab în LLC netratată anterior a fost evaluată într-un studiu randomizat, multicentric, deschis, de fază 3 (ELEVATE-TN) la 535 de pacienţi. Pacienţilor li s-a administrat Calquence în asociere cu obinutuzumab, Calquence în monoterapie sau obinutuzumab în asociere cu clorambucil. Studiul ELEVATE-TN a inclus pacienţi cu vârsta de 65 de ani ori peste sau cu vârsta între 18 şi 65 de ani şi afecţiuni medicale concomitente, iar 27,9% dintre pacienţi au avut valori ale ClCr < 60 ml/min. Dintre pacienţii cu vârsta < 65 de ani, 16,1% au avut un scor CIRS-G median de 8. Pacienţilor li s-a permis să administreze medicamente antitrombotice în cadrul studiului. Pacienţii care necesitau tratament anticoagulant cu warfarină sau antagonişti ai vitaminei K echivalenţi au fost excluşi.
Pacienţii au fost randomizaţi în raport de 1:1:1 în 3 braţe de tratament pentru a li se administra:
- Calquence în asociere cu obinutuzumab (Calquence+G): Calquence în doză de 100 mg a fost administrat de două ori pe zi începând din ziua 1 a ciclului 1, până la progresia bolii sau apariţia toxicităţii inacceptabile. Obinutuzumab a fost administrat începând din ziua 1 a ciclului 2 timp de maximum 6 cicluri de tratament. Obinutuzumab 1000 mg a fost administrat în zilele 1 şi 2 (100 mg în ziua 1 şi 900 mg în ziua 2), 8 şi 15 ale ciclului 2, iar ulterior în doză de 1000 mg în ziua 1 a ciclurilor 3-7. Fiecare ciclu a avut 28 de zile.
- Calquence în monoterapie: Calquence în doză de 100 mg a fost administrat de două ori pe zi până la progresia bolii sau apariţia toxicităţii inacceptabile.
- Obinutuzumab în asociere cu clorambucil (GClb): Obinutuzumab şi clorambucil au fost administrate timp de maximum 6 cicluri de tratament. Obinutuzumab 1000 mg a fost administrat în zilele 1 şi 2 (100 mg în ziua 1 şi 900 mg în ziua 2), 8 şi 15 ale ciclului 1, iar ulterior în doză de 1000 mg în ziua 1 a ciclurilor 2-6. Clorambucil în doză de 0,5 mg/kg a fost administrat în zilele 1 şi 15 ale ciclurilor 1-6. Fiecare ciclu a avut 28 de zile.
Pacienţii au fost stratificaţi în funcţie de statusul deleţiei 17p (prezenţă versus absenţă), statusul de performanţă ECOG (scor 0 sau 1 versus 2) şi regiunea geografică (America de Nord şi Europa Occidentală versus alte regiuni). După progresia confirmată a bolii, 45 de pacienţi randomizaţi în braţul cu GClb au trecut la tratamentul cu Calquence în monoterapie. Tabelul 6 prezintă rezumativ caracteristicile demografice şi caracteristicile bolii pentru populaţia de pacienţi a studiului la momentul iniţial.
Tabelul 6. Caracteristicile iniţiale ale pacienţilor (ELEVATE-TN) cu LLC netratată anterior
| Caracteristică | Calquence în asociere cu obinutuzumab N=179 | Calquence în monoterapie N=179 | Obinutuzumab în asociere cu clorambucil N=177 |
|---|---|---|---|
| Vârsta în ani; valoare mediană (interval) | 70 (41-88) | 70 (44-87) | 71 (46-91) |
| Sex masculin; % | 62 | 62 | 59,9 |
| Rasă caucaziană; % | 91,6 | 95 | 93,2 |
| Status de performanță ECOG 0-1; % | 94,4 | 92,2 | 94,4 |
| Intervalul median de timp de la diagnostic (luni) | 30,5 | 24,4 | 30,7 |
| Ganglioni voluminoşi cu dimensiunea ≥ 5 cm; % | 25,7 | 38 | 31,1 |
Categoria citogenetică/FISH; % Deleţia 17p Deleţia 11q Mutaţia TP53 IGHV fără mutaţii Cariotip complex (≥ 3 anomalii) | 9,5 17,3 11,7 57,5 16,2 | 8,9 17,3 10,6 66,5 17,3 | 9 18,6 11,9 65,5 18,1 |
| Stadiu Rai; % | |||
0 I II III IV | 1,7 30,2 20,1 26,8 21,2 | 0 26,8 24,6 27,9 20,7 | 0,6 28,2 27,1 22,6 21,5 |
Obiectivul principal a fost supraviețuirea fără progresia bolii (SFP) în braţul de tratament cu Calquence+G comparativ cu braţul tratat cu GClb, evaluată de o comisie independentă de evaluare (IRC) pe baza criteriilor din 2008 ale Grupului internaţional de lucru asupra leucemiei limfocitare cronice (International Workshop on Chronic Lymphocytic Leukaemia, IWCLL), cu includerea clarificării referitoare la limfocitoza asociată tratamentului (Cheson 2012). După o perioadă mediană de urmărire de 28,3 luni, SFP evaluată de IRC a indicat o reducere cu 90%, semnificativă statistic, a riscului de progresie a bolii sau deces pentru pacienţii cu LLC netratată anterior din braţul cu Calquence+G, comparativ cu pacienţii braţului cu GClb. Rezultatele cu privire la eficacitate sunt prezentate în tabelul 7.
Tabelul 7. Rezultatele privind eficacitatea conform evaluărilor IRC (ELEVATE-TN) la pacienţi cu
LLC
| Calquence în asociere cu obinutuzumab N=179 | Calquence în monoterapie N=179 | Obinutuzumab în asociere cu clorambucil N=177 | |
| Supraviețuirea fără progresia bolii* | |||
|---|---|---|---|
| Număr de evenimente (%) | 14 (7,8) | 26 (14,5) | 93 (52,5) |
| PB, n (%) | 9 (5) | 20 (11,2) | 82 (46,3) |
| Număr de decese (%) | 5 (2,8) | 6 (3,4) | 11 (6,2) |
| Valoare mediană (IÎ 95%), luni | NA | NA (34,2, NA) | 22,6 (20,2, 27,6) |
| RR† (IÎ 95%) | 0,10 (0,06, 0,17) | 0,20 (0,13, 0,30) | - |
| Valoare p | <0,0001 | <0,0001 | - |
| Estimare la 24 de luni, % (IÎ 95%) | 92,7 (87,4, 95,8) | 87,3 (80,9, 91,7) | 46,7 (38,5, 54,6) |
| Supravieţuirea generalăa | |||
| Număr de decese (%) | 9 (5) | 11 (6,1) | 17 (9,6) |
| Risc relativ (IÎ 95%) † | 0,47 (0,21, 1,06) | 0,60 (0,28, 1,27) | - |
| Rata celui mai bun răspuns general* (RC + RCi + RPn + RP) | |||
| RRG, n (%) (IÎ 95%) | 168 (93,9) (89,3; 96,5) | 153 (85,5) (79,6; 89,9) | 139 (78,5) (71,9; 83,9) |
| Valoare p | <0,0001 | 0,0763 | - |
| RC, n (%) | 23 (12,8) | 1 (0,6) | 8 (4,5) |
| RCi, n (%) | 1 (0,6) | 0 | 0 |
| RPn, n (%) | 1 (0,6) | 2 (1,1) | 3 (1,7) |
| RP, n (%) | 143 (79,9) | 150 (83,8) | 128 (72,3) |
IÎ = interval de încredere; RR = risc relativ; NA = nu a fost atinsă; RC = răspuns complet; RCi = răspuns complet cu recuperare hematologică incompletă; RPn = răspuns parţial nodular; RP = răspuns parţial;
*Conform evaluării de către IRC
†Pe baza modelului stratificat Cox al riscurilor proporţionale a Mediana SG nu a fost atinsă în ambele braţe de tratament.
Rezultatele privind SFP pentru Calquence în asociere cu sau fără obinutuzumab au fost consecvente la nivelul subgrupurilor, inclusiv al celor cu factori de risc înalt. La nivelul populaţiei de pacienţi cu LLC cu risc înalt (deleţie 17p, deleţie 11q, mutaţie TP53 sau IGHV fără mutaţii), RR aferent SFP pentru Calquence asociat sau nu cu obinutuzumab comparativ cu obinutuzumab plus clorambucil a fost de 0,08 [IÎ 95% (0,04, 0,15)] şi, respectiv, de 0,13 [IÎ 95% (0,08, 0,21)].
Tabelul 8. Analiza pe subgrupuri a SFP (studiul ELEVATE-TN)
| Calquence în monoterapie | Calquence+G | |||||
| N | Risc relativ | IÎ 95% | N | Risc relativ | IÎ 95% | |
| Toţi subiecţii | 179 | 0,20 | (0,13; 0,30) | 179 | 0,10 | (0,06; 0,17) |
Del 17p Da Nu | 19 160 | 0,20 0,20 | (0,06; 0,64) (0,12; 0,31) | 21 158 | 0,13 0,09 | (0,04; 0,46) (0,05; 0,17) |
Mutaţia TP53 Da Nu | 19 160 | 0,15 0,20 | (0,05; 0,46) (0,12; 0,32) | 21 158 | 0,04 0,11 | (0,01; 0,22) (0,06; 0,20) |
Del 17p sau/şi mutaţia TP53 Da Nu | 23 156 | 0,23 0,19 | (0,09; 0,61) (0,11; 0,31) | 25 154 | 0,10 0,10 | (0,03; 0,34) (0,05; 0,18) |
Status IGHV Cu mutaţii Fără mutaţii | 58 119 | 0,69 0,11 | (0,31; 1,56) (0,07; 0,19) | 74 103 | 0,15 0,08 | (0,04; 0,52) (0,04; 0,16) |
Del 11q Da Nu | 31 148 | 0,07 0,26 | (0,02; 0,22) (0,16; 0,41) | 31 148 | 0,09 0,10 | (0,03; 0,26) (0,05; 0,20) |
Cariotip complex Da Nu | 31 117 | 0,10 0,27 | (0,03; 0,33) (0,16; 0,46) | 29 126 | 0,09 0,11 | (0,03; 0,29) (0,05; 0,21) |
Conform datelor pe durată îndelungată, perioada mediană de urmărire a fost de 58,2 luni pentru brațul de tratament cu Calquence+G, 58,1 luni pentru brațul de tratament cu Calquence și 58,2 luni pentru brațul de tratament cu GClb. Mediana SFP evaluată de investigator nu a fost atinsă în braţele de tratament cu Calquence+G și Calquence în monoterapie şi a fost de 27,8 luni în braţul cu GClb. La momentul celei mai recente întreruperi a colectării datelor, un total de 72 de pacienți (40,7%) inițial randomizați în brațul cu GClb au trecut la tratamentul cu Calquence în monoterapie. Mediana supraviețuirii generale nu a fost atinsă în niciun braţ de tratament cu un total de 76 de decese: 18 (10,1%) în brațul cu Calquence+G, 30 (16,8%) în brațul cu Calquence în monoterapie și 28 (15,8%) în brațul cu GClb.
Tabelul 9. Rezultatele privind eficacitatea conform evaluărilor INV (ELEVATE-TN) la pacienţi cu
LLC
| Calquence în asociere cu obinutuzumab N=179 | Calquence în monoterapie N=179 | Obinutuzumab în asociere cu clorambucil N=177 | |
| Supraviețuirea fără progresia bolii | |||
|---|---|---|---|
| Număr de evenimente (%) | 27 (15,1) | 50 (27,9) | 124 (70,1) |
| PB, n (%) | 14 (7,8) | 30 (16,8) | 112 (63,3) |
| Număr de decese (%) | 13 (7,3) | 20 (11,2) | 12 (6,8) |
| Valoare mediană (IÎ 95%), luni* | NA | NA (66,5, NA) | 27,8 (22,6, 33,2) |
| RR† (IÎ 95%) | 0,11 (0,07, 0,16) | 0,21 (0,15, 0,30) | - |
| Supraviețuirea generală | |||
| Număr de decese (%) | 18 (10,1) | 30 (16,8) | 28 (15,8) |
| Risc relativ (IÎ 95%)† | 0,55 (0,30, 0,99) | 0,98 (0,58, 1,64) | - |
IÎ=interval de încredere; RR=risc relativ; NA=nu a fost atinsă
* 95% interval de încredere bazat pe estimarea Kaplan-Meier.
†Estimare bazată pe modelul stratificat Cox al riscurilor proporţionale pentru riscul relativ (IÎ 95%) stratificat în funcție de statusul deleției 17p (da sau nu).
Figura 1. Curba Kaplan-Meier a SFP evaluată de INV la pacienţii (ELEVATE-TN) cu LLC
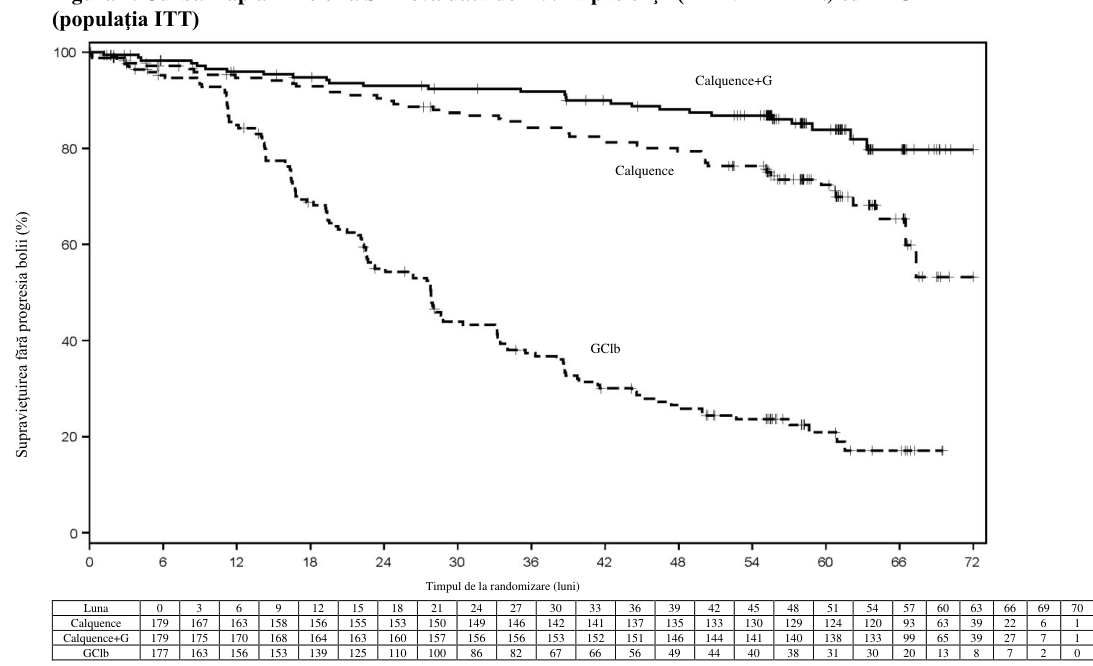
Pacienți cu LLC netratată anterior – terapie cu durată fixă
Calquence în asociere cu venetoclax cu sau fără obinutuzumab
Siguranța și eficacitatea Calquence în asociere cu venetoclax cu sau fără obinutuzumab în LLC netratată anterior au fost evaluate într-un studiu randomizat, multicentric, cu design deschis, de fază 3 (AMPLIFY) la 867 pacienți. Pacienților li s-a administrat Calquence în asociere cu venetoclax, Calquence în asociere cu venetoclax și obinutuzumab sau chimioimunoterapie la decizia medicului investigator, fie FCR (fludarabină plus ciclofosfamidă plus rituximab) sau BR (bendamustină plus rituximab). AMPLIFY a inclus pacienți cu LLC netratată anterior, fără mutații de tip deleție (17p) sau TP53, care au avut vârsta de 18 ani și peste. Studiul a permis ca pacienții să primească agenți antitrombotici, mai puțin warfarină sau alți antagoniști ai vitaminei K.
Pacienții au fost randomizați în raport de 1:1:1 în 3 brațe de tratament pentru a li se administra:
- Calquence plus venetoclax (AV): Calquence în doză de 100 mg a fost administrat de două ori pe zi începând cu ziua 1 a primului ciclu de tratament, pentru un număr total de 14 cicluri sau până la progresia bolii sau toxicitate inacceptabilă. În ziua 1 a ciclului 3, pacienții au inițiat programul de titrare a dozelor de venetoclax cu durata de 5 săptămâni, cu doze începând de la 20 mg, care au crescut săptămânal la 50 mg, 100 mg, 200 mg și, la final, 400 mg administrate o dată pe zi. Venetoclax a fost administrat pentru un număr total de 12 cicluri de tratament. Fiecare ciclu a avut
28 de zile.
- Calquence plus venetoclax plus obinutuzumab (AVO): Calquence în doză de 100 mg a fost administrat de două ori pe zi începând cu ziua 1 a primului ciclu de tratament, pentru un număr total de 14 cicluri sau până la progresia bolii sau toxicitate inacceptabilă. În ziua 1 a ciclului 3, pacienții au inițiat programul de titrare a dozelor de venetoclax cu durata de 5 săptămâni, cu doze începând de la 20 mg și care au crescut săptămânal la 50 mg, 100 mg, 200 mg și, la final, 400 mg administrate o dată pe zi. Venetoclax a fost administrat pentru un număr total de 12 cicluri de tratament. Obinutuzumab în doză de 1000 mg a fost administrat în ziua 1 sau în ziua 1 și 2 (100 mg în ziua 1 și 900 mg în ziua 1 sau 2), în zilele 8 și 15 ale ciclului 2, urmat de 1000 mg în ziua 1 a ciclurilor 3-7. Fiecare ciclu a avut 28 de zile.
- Chimioimunoterapie la decizia medicului investigator (FCR/BR):
- Fludarabină plus ciclofosfamidă plus rituximab (FCR): fludarabină (25 mg/m2) și ciclofosfamidă (250 mg/m2) au fost administrate în zilele 1-3, până la un maxim de 6 cicluri de tratament. Rituximab a fost administrat în doză de 375 mg/m2 în ziua 1 a ciclului 1 și 500 mg/m2 în ziua 1 a ciclurilor 2-6. Fiecare ciclu a avut 28 de zile.
- Bendamustină plus rituximab (BR): bendmaustină în doză de 90 mg/m2 a fost administrată în zilele 1 și 2, până la un maxim de 6 cicluri. Rituximab a fost administrat în doză de 375 mg/m2 în ziua 1 a ciclului 1 și 500 mg/m2 în ziua 1 a ciclurilor 2-6. Fiecare ciclu a avut 28 de zile.
Pacienții au fost stratificați în funcție de vârstă (> 65 de ani sau ≤ 65 de ani), de statusul mutațional IGHV (cu mutație sau fără mutație), de stadiul Rai (cu risc crescut [≥ 3] versus fără risc crescut) și de regiunea geografică (America de Nord și Europa de Vest versus altele). Tabelul 10 prezintă sumarul caracteristicilor demografice și clinice la momentul inițial pentru populația de pacienți din studiu.
Tabelul 10. Caracteristicile inițiale ale pacienților (AMPLIFY) cu LLC netratată anterior
| Caracteristică | AV N=291 | AVO N=286 | FCR/BR N=290 |
|---|---|---|---|
| Vârsta în ani; valoare mediană (interval) | 61 (31-84) | 61 (29-81) | 61 (26-86) |
| Sex masculin; % | 61,2 | 69,2 | 63,1 |
| Rasă caucaziană; % | 91,1 | 86,7 | 86,9 |
| Status de performanță ECOG 0-1; % | 90,0 | 95,1 | 90,3 |
| Intervalul median de timp de la diagnostic până la randomizare (luni) | 28,5 | 26,1 | 29,6 |
| Ganglioni voluminoși cu dimensiunea ≥5 cm; % | 38,8 | 35,0 | 42,8 |
| Categoria citogenetică/FISH; % | |||
| Deleție 11q | 17,5 | 19,6 | 15,9 |
| Cariotip complex (≥ 3 anomalii) | 15,5 | 16,1 | 14,5 |
| IGHV fără mutații; % | 57,4 | 59,1 | 59,3 |
| Stadiul Rai; % | |||
| 0 | 1,0 | 0,3 | 1,4 |
| I | 16,2 | 21,3 | 21,4 |
| II | 35,7 | 37,8 | 33,4 |
| III | 23,7 | 17,8 | 20,3 |
| IV | 23,4 | 22,7 | 23,4 |
Obiectivul principal a fost SFP în brațul de tratament AV comparativ cu chimioimunoterapia aleasă de medicul investigator (FCR/BR), evaluată conform IRC pe baza criteriilor din 2018 ale IWCLL. Alte obiective ale eficacității tratamentului au fost SFP în brațul de tratament AVO comparativ cu chimioimunoterapie aleasă de medicul investigator (FCR/BR) și SG atât în brațul de tratament AV comparativ cu chimioimunoterapie aleasă de medicul investigator (FCR/BR), cât și în brațul AVO comparativ cu chimioimunoterapie aleasă de medicul investigator (FCR/BR).
Rezultatele privind eficacitatea sunt prezentate în tabelul 11. Curba Kaplan-Meier a SFP-IRC este prezentată în figura 2.
Tabelul 11. Rezultatele privind eficacitatea (AMPLIFY) la pacienții cu LLC netratată anterior
| AV N=291 | AVO N=286 | FCR/BRa N=290 | |
| Supraviețuirea fără progresia bolii* | |||
|---|---|---|---|
| Număr de evenimente (%) | 89 (30,6) | 56 (19,6) | 95 (32,8) |
| PB, n (%) | 77 (26,5) | 23 (8,0) | 66 (22,8) |
| Număr de decese (%) | 12 (4,1) | 33 (11,5) | 29 (10,0) |
| Valoare mediană (IÎ 95%), luni | NC (51,1; NC) | NC (NC; NC) | 47,6 (43,3; NC) |
| RR† (IÎ 95%) | 0,65 (0,49; 0,87) | 0,42 (0,30; 0,59) | - |
| Valoarea p | 0,0038 | ˂0,0001 | - |
| Supraviețuirea globalăb | |||
| Număr de decese (%) | 23 (7,9) | 37 (12,9) | 44 (15,2) |
| RR† (IÎ 95%) | 0,42 (0,25; 0,70)c | 0,75 (0,48; 1,16) | - |
NC= Nu a putut fi calculat; IÎ= Interval de încredere; PB= Progresia bolii.
*Conform evaluării IRC.
†Conform modelului stratificat Cox al riscurilor proporționale pentru riscul relativ. aConform alegerii medicului investigator, 143 de pacienți au fost planificați să li se administreze FCR și 147 de pacienți să li se administreze BR. bDate privind SG la o perioadă de monitorizare suplimentară cu durata de 6 luni de la momentul analizei intermediare ale SFP. cValoarea p nu este semnificativă după ajustarea pentru multiplicitate.
Figura 2. Curba Kaplan-Meier a SFP evaluată conform IRC la pacienții (AMPLIFY) cu LLC netratată anterior (populaţia ITT)
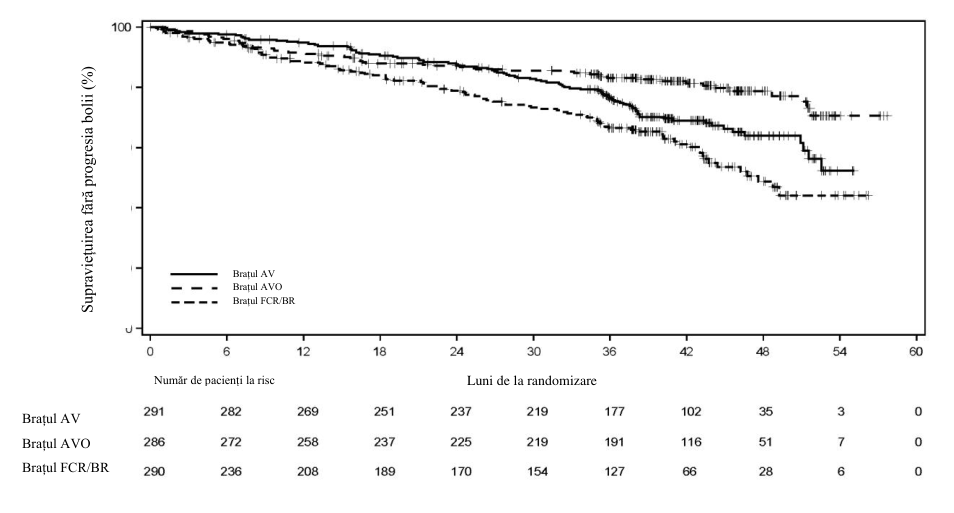
Pacienţi cu LLC cărora li s-a administrat cel puțin o terapie anterioară
Siguranţa şi eficacitatea Calquence în LLC recidivată sau refractară au fost evaluate într-un studiu randomizat, multicentric, deschis, de fază 3 (ASCEND) la 310 pacienţi cărora li se administrase minimum o terapie anterioară care nu includea inhibitori BCL-2 (ai limfomului cu celule B 2 mature) sau inhibitori ai receptorului celulelor B. Pacienţilor li s-a administrat Calquence în monoterapie sau terapia selectată de investigator fie idelalisib în asociere cu rituximab sau bendamustină în asociere cu rituximab. Pacienţilor li s-a permis să administreze medicamente antitrombotice în cadrul studiului. Pacienţii care necesitau tratament anticoagulant cu warfarină sau antagonişti ai vitaminei K echivalenţi au fost excluşi.
Pacienţii au fost randomizaţi în raport de 1:1 pentru a li se administra:
- Calquence în doză de 100 mg până la progresia bolii sau apariţia toxicităţii inacceptabile, sau
- La alegerea investigatorului:
- Idelalisib în doză de 150 mg de două ori pe zi în asociere cu rituximab în doză de 375 mg/m2 i.v. în ziua 1 a primului ciclu, apoi în doză de 500 mg/m2 i.v. la fiecare două săptămâni până la 4 doze, iar ulterior la intervale de 4 săptămâni până la 3 doze, administrându-se în total 8 perfuzii.
- Bendamustină în doză de 70 mg/m2 (zilele 1 şi 2 ale fiecărui ciclu de 28 de zile) în asociere cu rituximab (375 mg/m2/500 mg/m2) în ziua 1 a fiecărui ciclu de 28 de zile, timp de până la 6 cicluri.
Pacienţii au fost stratificaţi în funcţie de statusul deleţiei 17p (prezenţă versus absenţă), statusul de performanţă ECOG (scor 0 sau 1 versus 2) şi numărul terapiilor anterioare (1 până la 3 versus ≥ 4). După progresia confirmată a bolii, 35 de pacienţi randomizaţi la terapia aleasă de investigator fie idelalisib cu rituximab sau bendamustină cu rituximab au trecut la tratamentul cu Calquence. Tabelul 12 prezintă rezumativ caracteristicile demografice şi caracteristicile bolii pentru populaţia de pacienţi a studiului la momentul iniţial.
Tabelul 12. Caracteristicile iniţiale ale pacienţilor (ASCEND) cu LLC
| Caracteristică | Calquence în monoterapie N=155 | Terapia aleasă de investigator dintre idelalisib + rituximab şi bendamustină + rituximab N=155 |
|---|---|---|
| Vârsta în ani; valoare mediană (interval) | 68 (32-89) | 67 (34-90) |
| Sex masculin; % | 69,7 | 64,5 |
| Rasă caucaziană; % | 93,5 | 91,0 |
| Status de performanță ECOG; % | ||
| 0 | 37,4 | 35,5 |
| 1 | 50,3 | 51,0 |
| 2 | 12,3 | 13,5 |
| Intervalul median de timp de la diagnostic (luni) | 85,3 | 79,0 |
| Ganglioni voluminoşi cu dimensiunea ≥ 5 cm; % | 49,0 | 48,4 |
Numărul median de terapii anterioare pentru LLC (interval) | 1 (1-8) | 2 (1-10) |
Numărul terapiilor anterioare pentru LLC; % 1 2 3 ≥ 4 | 52,9 25,8 11,0 10,3 | 43,2 29,7 15,5 11,6 |
| Categoria citogenetică/FISH; % | ||
| Deleţia 17p | 18,1 | 13,5 |
| Deleţia 11q | 25,2 | 28,4 |
| Mutaţia TP53 | 25,2 | 21,9 |
| IGHV fără mutaţii | 76,1 | 80,6 |
| Cariotip complex (≥ 3 anomalii) | 32,3 | 29,7 |
| Stadiu Rai; % | ||
| 0 | 1,3 | 2,6 |
| I | 25,2 | 20,6 |
| II | 31,6 | 34,8 |
| III | 13,5 | 11,6 |
| IV | 28,4 | 29,7 |
Obiectivul principal a fost SFP determinată de IRC pe baza criteriilor IWCLL 2008, cu includerea clarificării referitoare la limfocitoza asociată tratamentului (Cheson 2012). După o perioadă de urmărire mediană de 16,1 luni, SFP a indicat o reducere cu 69%, semnificativă statistic, a riscului de deces sau progresie a bolii pentru pacienţii din braţul de tratament cu Calquence. Rezultatele cu privire la eficacitate sunt prezentate în tabelul 13. Curba Kaplan-Meier pentru SFP este prezentată în Figura 3.
Tabelul 13. Rezultatele privind eficacitatea conform evaluărilor IRC (ASCEND) la pacienţi cu LLC
| Calquence în monoterapie N=155 | Terapia aleasă de investigator fie idelalisib + rituximab sau bendamustină + rituximab N=155 | |
| Supraviețuirea fără progresia bolii | ||
|---|---|---|
| Număr de evenimente (%) | 27(17,4) | 68 (43,9) |
| PB, n (%) | 19 (12,3) | 59 (38,1) |
| Număr de decese (%) | 8 (5.2) | 9 (5,8) |
| Valoare mediană (IÎ 95%), luni | NA | 16,5 (14,0, 17,1) |
| RR† (IÎ 95%) | 0,31 (0,20, 0,49) | |
| Valoare p | <0,0001 | |
| Estimare la 15 de luni, % (IÎ 95%) | 82,6 (75,0, 88,1) | 54,9 (45,4, 63,5) |
| Supravieţuirea generalăa | ||
| Număr de decese (%) | 15 (9,7) | 18 (11,6) |
| Risc relativ (IÎ 95%) † | 0,84 (0,42, 1,66) | - |
| Rata celui mai bun răspuns general* (RC + RCi + RPn + RP)** | ||
| RRG, n (%) (IÎ 95%) | 126 (81,3) (74,4, 86,6) | 117 (75,5) (68,1; 81,6) |
| Valoare p | 0,2248 | - |
| RC, n (%) | 0 | 2 (1,3) |
| RP, n (%) | 126 (81,3) | 115 (74,2) |
| Durata răspunsului (DR) | ||
| Valoare mediană (IÎ 95%), luni | NA | 13,6 (11,9, NA) |
IÎ = interval de încredere; RR = risc relativ; NA = nu a fost atinsă; RC = răspuns complet; RCi = răspuns complet cu recuperare hematologică incompletă; RPn = răspuns parţial nodular; RP = răspuns parţial; PB = progresia bolii *Conform evaluării IRC.
aMediana SG nu a fost atinsă în ambele braţe de tratament. P<0,6089 pentru SG.
**RCi şi RPn au valori de 0.
†Pe baza modelului stratificat Cox al riscurilor proporţionale.
Figura 3. Curba Kaplan-Meier a SFP evaluată de IRC la pacienţii (ASCEND) cu LLC (populaţia
ITT)
| Calquence Terapia aleasă de investigator | |||||||||||||||||||||||||
100
80.
Supravieţuirea fără progresia bolii (%)
60.
40.
20.
0. 0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23
Timpul de la randomizare (luni)
| Număr de pacienți la risc | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Luna | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | 23 |
| Calquence | 155 | 153 | 153 | 149 | 147 | 146 | 145 | 143 | 143 | 139 | 139 | 137 | 118 | 116 | 73 | 61 | 60 | 25 | 21 | 21 | 1 | 1 | 1 | 0 |
| Terapia aleasă de investigator | 155 | 150 | 150 | 146 | 144 | 142 | 136 | 130 | 129 | 112 | 105 | 101 | 82 | 77 | 56 | 44 | 39 | 18 | 10 | 8 | 0 | |||
Rezultatele privind SFP pentru Calquence au fost consecvente la nivelul subgrupurilor, inclusiv al celor cu factori de risc înalt. La nivelul populaţiei cu LLC cu risc înalt (deleţie 17p, deleţie 11q, mutaţie TP53 şi IGHV fără mutaţii), RR pentru SFP a fost de 0,27 [IÎ 95% (0,17, 0,44)].
Tabelul 14. Analiza pe subgrupuri a SFP conform evaluării IRC (studiul ASCEND)
| Calquence în monoterapie | |||
| N | Risc relativ | IÎ 95% | |
| Toţi subiecţii | 155 | 0,30 | (0,19; 0,48) |
Del 17p Da Nu | 28 127 | 0,21 0,33 | (0,07; 0,68) (0,21; 0,54) |
Mutaţia TP53 Da Nu | 39 113 | 0,24 0,33 | (0,11; 0,56) (0,20; 0,57) |
Del 17p sau mutaţie TP53 Da Nu | 45 108 | 0,21 0,36 | (0,09; 0,48) (0,21; 0,61) |
| Status IGHV Cu mutaţii Fără mutaţii | 33 118 | 0,32 0,32 | (0,11; 0,94) (0,19; 0,52) |
Del 11q Da Nu | 39 116 | 0,28 0,31 | (0,11; 0,70) (0,19; 0,53) |
Cariotip complex Da Nu | 50 97 | 0,32 0,23 | (0,16; 0,63) (0,12; 0,44) |
Conform evaluării investigatorului la analiza finală, cu o perioadă mediană de urmărire de 46,5 luni pentru Calquence şi de 45,3 luni pentru IR/BR, a fost observată o reducere cu 72% a riscului de progresie a bolii sau deces pentru pacienții din brațul de tratament cu Calquence. Mediana SFP evaluată de investigator nu a fost atinsă în braţul de tratament cu Calquence şi a fost de 16,8 luni în braţul cu IR/BR. Rezultatele cu privire la eficacitate conform evaluărilor investigatorului (INV) sunt prezentate în tabelul 15. Curba Kaplan-Meier pentru SFP conform evaluării INV este prezentată în Figura 4.
Tabelul 15. Rezultatele privind eficacitatea la analiza finală, conform evaluărilor INV la pacienţii
(ASCEND) cu LLC
| Calquence în monoterapie N=155 | Terapia aleasă de investigator fie idelalisib + rituximab sau bendamustină + rituximab N=155 | |
| Supraviețuirea fără progresia bolii* | ||
|---|---|---|
| Număr de evenimente (%) | 62 (40,0) | 119 (76,8) |
| PB, n (%) | 43 (27,7) | 102 (65,8) |
| Număr de decese (%) | 19 (12,3) | 17 (11,0) |
| Valoare mediană (IÎ 95%), luni | NA | 16,8 (14,1, 22,5) |
| RR† (IÎ 95%) | 0,28 (0,20, 0,38) | |
| Supravieţuirea generalăa | ||
| Număr de decese (%) | 41 (26,5) | 54 (34,8) |
| Risc relativ (IÎ 95%) † | 0,69 (0,46, 1,04) | - |
IÎ = interval de încredere; RR = risc relativ; NA = nu a fost atinsă; PB = progresia bolii
*Conform evaluării INV. aMediana SG nu a fost atinsă în ambele braţe de tratament. P=0,0783 pentru SG.
†Pe baza modelului stratificat Cox al riscurilor proporţionale.
Figura 4. Curba Kaplan-Meier a SFP evaluată de INV la analiza finală, la pacienţii (ASCEND) cu
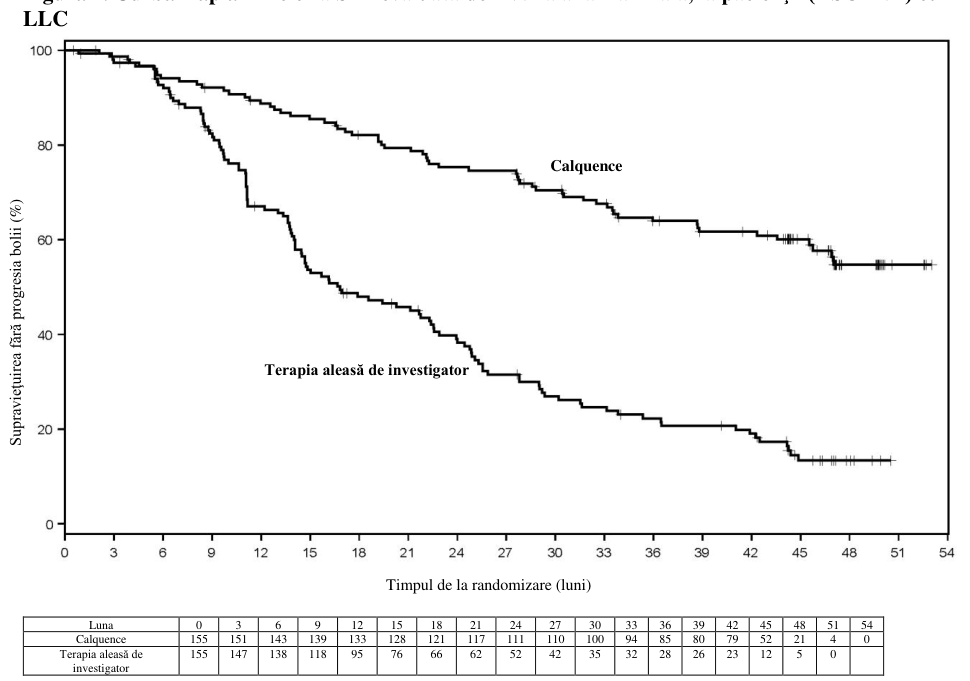
Rezultatele privind SFP pentru Calquence evaluate de investigator la analiza finală au fost consecvente la nivelul subgrupurilor, inclusiv al celor cu factori de risc înalt și au fost în concordanță cu analiza primară.
Pacienți cu LCM fără tratament anterior
Siguranța și eficacitatea Calquence la pacienții cu LCM fără tratament anterior au fost evaluate în studiul ECHO, un studiu randomizat, de fază III, multicentric, dublu-orb, controlat cu placebo. Au fost incluși 598 de pacienți cu vârsta de 65 de ani și peste cu diagnostic confirmat de LCM, fără tratament anterior. Pacienții au fost randomizați în raport 1:1 în două brațe de tratament pentru a li se administra:
- Calquence în asociere cu bendamustină și rituximab (Calquence + BR): s-a administrat Calquence în doze de 100 mg de două ori pe zi în mod continuu începând cu ziua 1 a primului ciclu de tratament. A fost administrată bendamustină în doze de 90 mg/m2 intravenos timp de 30 minute în zilele 1 și 2 ale fiecărui ciclu de tratament cu durata de 28 de zile, pentru un total de șase cicluri; și rituximab în doze de 375 mg/m2 intravenos în ziua 1 a fiecărui ciclu de tratament cu durata de 28 de zile, pentru un total de șase cicluri. Calquence + BR a fost administrat pentru maxim 6 cicluri de tratament (tratament de inducție).
- Placebo în asociere cu bendamustină și rituximab (placebo + BR): s-a administrat placebo de două ori pe zi în mod continuu începând cu ziua 1 a primului ciclu de tratament. A fost administrată bendamustină în doze de 90 mg/m2 intravenos timp de 30 minute în zilele 1 și 2 ale fiecărui ciclu de tratament cu durata de 28 de zile, pentru un total de șase cicluri; și rituximab în doze de 375 mg/m2 intravenos în ziua 1 a fiecărui ciclu de tratament cu durata de 28 de zile, pentru un total de șase cicluri. Placebo + BR a fost administrat pentru maxim de 6 cicluri de tratament (tratament de inducție).
S-a administrat Calquence sau placebo în mod continuu până la progresia bolii sau până la apariția toxicității inacceptabile. După tratamentul de inducție, pacienții care au obținut răspuns (RP sau RC) au primit tratament de menținere cu rituximab în doze de 375 mg/m2 în ziua 1 a fiecărui al doilea ciclu, până la 12 doze suplimentare, până la ciclul 30 de tratament. Pacienții randomizați pentru a li se administra placebo + BR, la care s-a raportat progresia bolii au fost eligibili pentru a face trecerea în brațul cu Calquence ca monoterapie în doze de 100 mg de două ori pe zi, până la a doua progresie a bolii sau până la apariția toxicității inacceptabile.
Procesul de randomizare a pacienților a fost stratificat în funcție de regiunea geografică (America de Nord versus Europa de Vest versus Altele) și de scorul Indicelui de Prognostic Internațional simplificat pentru LCM (Mantle Cell Lymphoma International Prognostic Index, MIPI) (0-3 versus 4-5 versus 6-11).
Vârsta mediană a pacienților a fost de 71 de ani (65-86), 70,7 % au fost bărbați, 78,3% au fost caucazieni și 93,1% au avut status de performanță ECOG 0-1. Scorul simplificat MIPI a fost scăzut (0-3) la 33,1% dintre pacienți, intermediar (4-5) la 42,8% dintre pacienți și crescut (6-11) la 24,1% dintre pacienți. În total, 37,7% dintre pacienți au prezentat masă tumorală ≥ 5 cm și 86% au prezentat boală în stadiul IV conform clasificării Ann Arbor. Au fost raportate variante agresive ale LCM, precum formele blastoide și pleomorfice la 7,7% și 5,5% dintre pacienți. În total, 47,8% dintre pacienți au avut scor Ki-67 ≥ 30%. Caracteristicile inițiale au fost similare în cele două brațe de tratament.
Obiectivul primar a fost supraviețuirea fără progresia bolii (SFP), evaluată de IRC conform Clasificării Lugano 2014 pentru limfom non-Hodkin (LNH) la pacienții cu LCM netratat anterior. Suplimentar, a fost evaluată de IRC și rata de răspuns global (RRG).
SFP a fost evaluată de IRC la o perioadă mediană de urmărire de 49,8 luni.
Supraviețuirea globală mediană nu a fost atinsă în niciun braț de tratament, cu o perioadă de urmărire suplimentară de 6 luni de la analiza primară a SFP și cu o urmărire mediană de 63,0 luni. Au fost raportate 218 decese: 105 (35,1%) în grupul tratat cu Calquence + BR și 113 (37,8%) în grupul cu administrare de placebo + BR. Rezultatele de eficacitate sunt prezentate în tabelul 16. Curbele Kaplan-Meier ale SFP sunt prezentate în figura 5.
Tabelul 16. Rezultate privind eficacitatea la pacienții fără tratament anterior pentru LCM din studiul ECHO
| Calquence + BR N=299 | Placebo + BR N=299 | |
| SFP evaluată conform IRC | ||
|---|---|---|
| Mediana (IÎ 95%) | 66,4 (55,1, NE) | 49,6 (36,0, 64,1) |
| RR (IÎ 95%) (stratificată)* | 0,73 (0,57, 0,94) | |
| Valorea p‡ | 0,0160 | |
| RRG evaluată conform IRC | ||
| RC + RP n (%) | 272 (91,0) | 263 (88,0) |
| IÎ 95% | 87,3, 93,8 | 83,9, 91,3 |
| RC n (%) | 199 (66,6) | 160 (53,5) |
| RP n (%) | 73 (24,4) | 103 (34,4) |
| Valoarea p | 0,2196 | - |
RR = risc relativ, RC = răspuns complet, RP = răspuns parțial, NE = neevaluabil
*Stratificat pe baza factorilor de randomizarea: regiune geografică (America de Nord, Europa de Vest, altele) și scorul simplificat MIPI (risc scăzut [între 0 și 3], risc intermediar [între 4 și 5], risc crescut [între 6 și 11], datele fiind colectate cu ajutorul IXRS.
Estimat pe baza modelului stratificat Cox al riscurilor proporționale pentru evaluarea ratei de risc (IÎ 95%). ‡Estimat pe baza testului stratificat log-rank pentru valoarea P.
Figura 5. Curba Kaplan-Meier a SFP evaluată de IRC la pacienții cu LCM fără tratament anterior
(ECHO)
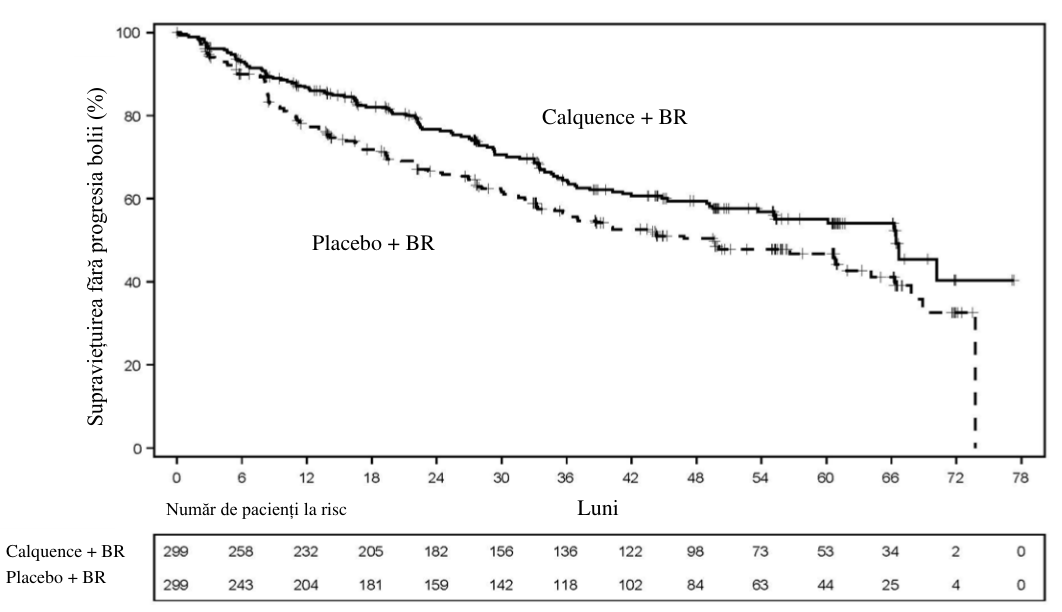
Pacienţi cu LCM cărora li s-a administrat cel puțin o terapie anterioară
Siguranța și eficacitatea CALQUENCE la pacienții cu LCM au fost evaluate în cadrul unui studiu de fază II, deschis, multicentric, cu un singur braț de tratament (ACE-LY-004) care a inclus 124 pacienți tratați anterior. Tuturor pacienţilor li s-a administrat CALQUENCE pe cale orală, în doză de 100 mg de două ori pe zi până la progresia bolii sau toxicitate inacceptabilă. Studiul nu a inclus pacienții cărora li se administrase tratament anterior cu inhibitori TKB sau BCL-2. Obiectivul principal a fost rata răspunsului general (RRG) evaluată de investigator conform clasificării Lugano pentru limfomul non-Hodgkin (non- Hodgkin’s lymphoma, LNH). Un obiectiv suplimentar de evaluare a fost durata răspunsului (DR). Rezultatele eficacității la analiza finală (54 de luni) sunt prezentate în tabelul 17.
La analiza finală, vârsta mediană a fost de 68 de ani (interval 42 – 90 de ani), 79,8% erau bărbați, iar 74,2% erau caucazieni. La momentul inițial, 92,8% dintre pacienți aveau un status al performanței ECOG 0 sau 1. Timpul median de la diagnostic a fost de 46,3 luni iar numărul median de tratamente anterioare a fost de 2 (interval 1 - 5), incluzând cei 17,7% dintre pacienții cu transplant anterior de celule stem. Cele mai frecvente tratamente anterioare au avut la bază schema terapeutică CHOP (51,6%) și citarabina (ARA-C) (33,9%). La momentul inițial, 37,1% dintre pacienți aveau cel puțin o tumoră cu diametrul cel mai mare ≥ 5 cm, iar 72,6% aveau implicare extraganglionară, incluzând cei 50,8% dintre pacienții cu afectare a măduvei osoase. Scorul MIPI simplificat (care include vârsta, scorul ECOG și valorile inițiale de lactat dehidrogenază și ale numărului de leucocite) a fost intermediar la 43,5% dintre pacienți și ridicat la 16,9% dintre pacienți.
Tabelul 17. RRG și DR la pacienții cu LCM din studiul ACE-LY-004 la analiza finală de 54 luni
| Evaluarea investigatorului la 54 luni N=124 n (%) (IÎ 95%*) | |
| Rata răspunsului general (RRG) | |
|---|---|
| Rata răspunsului general | 101 (81,5%) (73,5; 87,9) |
| Răspuns complet | 59 (47,6%) (38,5; 56,7) |
| Răspuns parțial | 42 (33,9%) (25,6; 42,9) |
| Nu poate fi estimatㆠ| 3 (2,4%) (0,5; 6,9) |
| Durata răspunsului (DR) | |
| Valoarea mediană (luni) | 28,6 (17,5; 39,1) |
IÎ=interval de încredere *95% interval de încredere binomial exact. †Include subiecți fără nicio evaluare adecvată a bolii după momentul inițial. | |
Populația pediatrică
Agenţia Europeană a Medicamentului a acordat o derogare de la obligaţia de depunere a rezultatelor studiilor efectuate cu Calquence la toate subgrupele de copii şi adolescenţi pentru tratamentul neoplasmelor cu celule B mature (vezi pct. 4.2 pentru informaţii privind utilizarea la copii şi adolescenţi).
5.2 Proprietăţi farmacocinetice
Farmacocinetica (FC) acalabrutinib şi a metabolitului său activ, ACP-5862, au fost studiate la subiecţi sănătoşi şi la pacienţi cu afecțiuni maligne cu celule B. Expunerea la acalabrutinib este proporţională cu doza şi atât acalabrutinib, cât şi ACP-5862 au o FC aproape liniară la administrarea unor doze cuprinse între 75 şi 250 mg. Modelarea FC populaţională sugerează că farmacocinetica acalabrutinib şi a ACP-5862 este similară la pacienţi cu afecțiuni maligne cu celule B diferite. La doza recomandată de 100 mg de două ori pe zi administrată pacienţilor cu afecțiuni maligne cu celule B (inclusiv LLC), media geometrică a concentraţiilor plasmatice zilnice la starea de echilibru în funcţie de timp (ASC24ore) şi concentraţia plasmatică maximă (Cmax) pentru acalabrutinib au fost de 1679 ngxoră/ml şi, respectiv, de 438 ng/ml, iar pentru ACP-5862 au fost de 4166 ngxoră/ml şi, respectiv, de 446 ng/ml.
S-a demonstrat bioechivalența între Calquence sub formă de comprimate și Calquence sub formă de capsule. Calquence sub formă de comprimate conține acalabrutinib maleat, o formă de sare a acalabrutinib, care prezintă o solubilitate mai mare în condiții de pH crescut, în comparație cu acalabrutinib sub formă de bază, care constituie substanța activă din Calquence capsule. Calquence sub formă de comprimate prezintă astfel o absorbție mai bună atunci când este administrat concomitent cu medicamente care reduc secreția de acid gastric.
Absorbție
Timpul până la atingerea concentraţiilor plasmatice maxime (Tmax) a fost de 0,2-3,0 ore pentru acalabrutinib şi de 0,5-4,0 ore pentru ACP-5862. Biodisponibilitatea absolută a Calquence a fost de 25%.
Efectul alimentelor asupra acalabrutinib
La voluntarii sănătoşi, administrarea unei singure doze de 100 mg de acalabrutinib sub formă de comprimate în timpul unei mese hipercalorice şi cu conţinut ridicat de grăsimi (aproximativ 918 calorii, 59 grame carbohidraţi, 59 grame lipide şi 39 grame proteine) nu a influenţat valorile medii ale ASC comparativ cu administrarea în condiţii de repaus alimentar. Valorile Cmax rezultate au scăzut cu 54% şi Tmax a fost întârziat cu 1-2 ore.
Distribuție
Proporţia legării reversibile de proteinele plasmatice umane a fost de 99,4% pentru acalabrutinib şi de 98,8% pentru ACP-5862. Raportul mediu sânge-plasmă in vitro a fost de 0,8 pentru acalabrutinib şi de 0,7 pentru ACP-5862. Volumul de distribuţie mediu la starea de echilibru (Vss) a fost de aproximativ 34 l pentru acalabrutinib.
Biotransformare/metabolizare
In vitro, acalabrutinib este predominant metabolizat de enzimele CYP3A şi în proporţie minoră prin conjugarea cu glutation şi hidroliză amidică. ACP-5862 a fost identificat ca metabolit principal în plasmă, care ulterior a fost metabolizat în special prin oxidare mediată de CYP3A, cu o medie geometrică a expunerii (ASC) de aproximativ 2 până la 3 ori mai mare decât expunerea la acalabrutinib. ACP-5862 are o putere de inhibiţie a TKB cu aproximativ 50% mai mică decât cea a acalabrutinib.
Studiile in vitro indică faptul că acalabrutinib nu inhibă CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, UGT1A1 sau UGT2B7 la concentraţii relevante clinic şi probabilitatea ca acesta să afecteze clearance-ul medicamentelor substrat pentru aceste enzime ale CYP este foarte redusă.
Studiile in vitro indică faptul că ACP-5862 nu inhibă CYP1A2 , CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4/5, UGT1A1 sau UGT2B7 la concentraţii relevante clinic şi probabilitatea ca acesta să afecteze clearance-ul medicamentelor substrat pentru aceste enzime ale CYP este foarte redusă.
Interacţiuni cu proteinele transportoare
Studiile in vitro indică faptul că acalabrutinib şi ACP-5862 sunt substraturi pentru gp-P şi BCRP. Cu toate acestea, este improbabil ca administrarea concomitent cu inhibitori ai BCRP să determine interacţiuni medicamentoase relevante din punct de vedere clinic. Administrarea concomitent cu un inhibitor al OATP1B1/1B3 (rifampicină în doză unică de 600 mg) a determinat o creştere a valorilor Cmax şi ASC ale acalabrutinib de 1,2 ori şi, respectiv, de 1,4 ori (N=24, subiecţi sănătoşi), ceea ce nu este relevant din punct de vedere clinic.
La administrarea în concentraţii relevante clinic, acalabrutinib şi ACP-5862 nu inhibă gp-P, OAT1, OAT3, OCT2, OATP1B1, OATP1B3 şi MATE2-K. Acalabrutinib poate inhiba BCRP de la nivel intestinal, în timp ce ACP-5862 poate inhiba MATE1 la administrarea în concentraţii relevante clinic (vezi pct. 4.5). Acalabrutinib nu inhibă MATE1, iar ACP-5862 nu inhibă BCRP la administrarea în concentraţii relevante clinic.
Eliminare
După administrarea unei singure doze orale de 100 mg de acalabrutinib sub formă de comprimate, media geometrică a timpului terminal de înjumătăţire plasmatică prin eliminare (t1/2) pentru acalabrutinib a fost de 1,4 ore. Pentru metabolitul activ ACP-5862, t1/2 a fost de 6,6 ore.
Valoarea medie a clearance-ului oral aparent (CL/F) a fost de 134 l/oră pentru acalabrutinib şi de 22 l/oră pentru ACP-5862 la toţi pacienţii cu afecțiuni maligne cu celule B.
După administrarea unei singure doze de 100 mg de acalabrutinib marcate radioactiv [14C] la voluntari sănătoşi, 84% din doza administrată a fost recuperată din materiile fecale şi 12% din doză a fost recuperată din urină, mai puţin de 2% din doză fiind excretată sub formă de acalabrutinib nemodificat.
Grupe speciale de pacienți
Pe baza analizei farmacocinetice populaţionale, vârsta (>18 ani), sexul, rasa (caucaziană, afro-americană) şi greutatea corporală nu au avut efecte relevante clinic asupra farmacocineticii acalabrutinib şi a metabolitului său activ, ACP-5862.
Copii și adolescenți
Nu s-au efectuat studii farmacocinetice cu Calquence la pacienţi cu vârsta sub 18 ani.
Insuficienţă renală
Acalabrutinib este eliminat în proporţie minimală pe cale renală. Nu s-a efectuat niciun studiu farmacocinetic la pacienţi cu insuficiență renală.
Pe baza analizei FC populaţionale, nu au fost observate diferenţe relevante din punct de vedere farmacocinetic între cei 408 de subiecţi cu insuficienţă renală uşoară (RFGe între 60 şi 89 ml/min/1,73 m2, conform estimării prin ecuaţia MDRD), cei 109 subiecţi cu insuficiență renală moderată (RFGe între 30 şi 59 ml/min/1,73 m2) şi cei 192 de subiecţi cu funcţie renală în parametri normali (RFGe mai mare sau egală cu 90 ml/min/1,73 m2). Farmacocinetica acalabrutinib nu a fost caracterizată la pacienţi cu insuficiență renală severă (RFGe sub 29 ml/min/1,73 m2) sau la cei cu insuficiență renală necesitând tratament prin dializă. Pacienţii cu niveluri ale creatininei de 2,5 ori mai mari decât LSVN instituţională nu au fost incluşi în studiile clinice (vezi pct. 4.2).
Insuficiență hepatică
Acalabrutinib este metabolizat pe cale hepatică. În studiile dedicate efectuate la pacienţi cu insuficiență hepatică (IH) comparativ cu pacienţi cu funcţie hepatică normală (N=6), expunerea la acalabrutinib (ASC) a crescut de 1,9 ori, 1,5 ori şi de 5,3 ori la pacienţii cu insuficiență hepatică uşoară (N=6) (Clasificarea Child-Pugh clasa A), moderată (N=6) (Clasificarea Child-Pugh clasa B) şi, respectiv, severă (N=8) (Clasificarea Child-Pugh clasa C). La subiecţii din grupul cu IH moderată, modificările markerilor relevanţi pentru capacitatea de eliminare a medicamentelor nu au fost însă semnificative, prin urmare este posibil ca efectul insuficienţei hepatice moderate să fi fost subestimat în cadrul acestui studiu. Pe baza analizei farmacocinetice populaţionale, nu au fost observate diferenţe relevante clinic între subiecţii cu insuficienţă hepatică uşoară (N=79) sau moderată (N=6) (valori ale bilirubinei totale de 1,5 până la 3 ori mai mari decât LSVN şi orice valori ale AST) şi cei cu funcţie hepatică normală (N=613) (valori ale bilirubinei totale şi AST în limitele LSVN) (vezi pct. 4.2).
5.3 Date preclinice de siguranţă
Carcinogenicitate
Nu s-au efectuat studii de carcinogenicitate cu acalabrutinib.
Genotoxicitate/mutagenicitate/fototoxicitate
Acalabrutinib nu a fost mutagen în cadrul testului mutaţiei inverse bacteriene, al testului in vitro pentru detectarea aberaţiilor cromozomiale sau al testului in vivo al micronucleilor de la nivelul măduvei hematogene la şoarece.
Pe baza testelor de fototoxicitate in vitro pe linii celulare 3T3, se consideră că acalabrutinib prezintă un risc scăzut de fototoxicitate la om.
Toxicitatea după doze repetate
La şobolani au fost observate modificări microscopice la nivelul pancreasului, minimale până la uşoare ca severitate (hemoragie/pigmenţi/inflamaţie/fibroză în insulele pancreatice) la toate dozele testate. Au fost observate modificări non-adverse, minimale până la uşoare ca severitate, la nivelul rinichilor (bazofilie tubulară, regenerare tubulară şi inflamaţie) în studii cu durata de până la 6 luni în care s-a administrat la şobolani doza maximă fără efecte adverse observabile (No Observed Adverse Effect Level, NOAEL) de 30 mg/kg şi zi. Expunerile medii (ASC) în cazul administrării NOAEL la şobolani masculi şi femele corespund unui nivel de 0,6 ori şi respectiv 1,0 ori expunerea clinică la doza recomandată de 100 mg de două ori pe zi. Doza minimă cu efecte adverse observabile (Lowest Adverse Observed Effect Level, LOAEL) la care au fost înregistrate modificări reversibile la nivel renal (degenerare tubulară moderată) şi hepatic (necroza hepatocitelor individuale) în studiul pe termen lung la şobolani a fost de 100 mg/kg şi zi, şi a determinat o expunere de 4,2 ori mai mare decât expunerea cu doza recomandată de 100 mg de două ori pe zi. În studii cu durata de 9 luni efectuate la câini, NOAEL a fost de 10 mg/kg şi zi, ceea ce corespunde unei expuneri de 3 ori mai mare decât ASC la doza clinică recomandată. La administrarea unei doze de 30 mg/kg şi zi la câini (de 9 ori mai mare decât ASC clinică) au fost observate degenerări tubulare minimale la nivelul rinichilor, scăderi uşoare ale greutăţii splinei, scăderi tranzitorii, minimale până la uşoare, ale masei eritrocitare şi creşteri ale ALT şi ALP. Toxicităţile cardiace la şobolani (hemoragie, inflamație, necroză miocardică) şi câini (inflamaţie perivasculară/vasculară) au fost înregistrate numai la animale decedate pe parcursul studiilor, în contextul administrării de doze mai mari decât doza maximă tolerată (DMT). Expunerile la şobolani şi câini asociate cu modificări cardiace au fost cel puţin de 6,8 ori şi, respectiv, de 25 ori mai mari decât ASC clinică. Reversibilitatea acestor modificări cardiace nu a putut fi evaluată deoarece acestea au fost observate numai la administrarea unor doze peste DMT.
Efectele toxice asupra funcţiei de reproducere
Nu au fost observate efecte asupra fertilităţii la şobolanii masculi sau femele la expuneri de 10 sau, respectiv, 9 ori mai mari decât ASC clinică la doza recomandată.
Nu au fost observate efecte asupra dezvoltării şi supravieţuirii embrio-fetale la femelele de şobolan gestante, în cazul unor expuneri de aproximativ 9 ori mai mari decât ASC observată la pacienţii trataţi cu doza recomandată de 100 mg de două ori pe zi. În două studii de evaluare a efectelor asupra reproducerii la şobolani, au fost observate cazuri de distocie (travaliu dificil/prelungit) la expuneri de > 2,3 ori mai mari decât expunerea clinică la doza de 100 mg de două ori pe zi. S-a confirmat prezenţa acalabrutinib şi a metabolitului său activ în plasma fetuşilor de şobolan. Prezenţa acalabrutinib şi a metabolitului său activ a fost depistată în laptele matern al femelelor de şobolan.
Într-un studiu de toxicitate embrio-fetală pe femele de iepure gestante au fost observate scăderi ale greutăţii fetuşilor şi întârzieri ale osificării la niveluri de expunere asociate cu toxicitate maternă care au fost de 2,4 ori mai mari decât valorile ASC la om atunci când s-a administrat doza recomandată.
6. PROPRIETĂŢI FARMACEUTICE
6.1 Lista excipienţilor
Nucleul comprimatului
Manitol (E421)
Celuloză microcristalină (E460)
Hidroxipropilceluloză de joasă substituție (E463)
Stearil fumarat de sodiu
Filmul comprimatului
Hipromeloză (E464)
Copovidonă
Dioxid de titan (E171)
Macrogol
Trigliceride cu lanț mediu
Oxid galben de fer (E172)
Oxid roșu de fer (E172)
6.2 Incompatibilităţi
Nu este cazul.
6.3 Perioada de valabilitate
3 ani.
6.4 Precauţii speciale pentru păstrare
Acest medicament nu necesită condiţii speciale de păstrare.
6.5 Natura şi conţinutul ambalajului
Ambalaje cu blistere din aluminiu/aluminiu, cu simbolurile soarelui/lunii, care conţin 8 sau 10 comprimate filmate. Cutii a câte 56 sau 60 de comprimate.
Este posibil ca nu toate mărimile de ambalaj să fie comercializate.
6.6 Precauţii speciale pentru eliminare
Orice medicament neutilizat sau material rezidual trebuie eliminat în conformitate cu reglementările locale.
7. DEŢINĂTORUL AUTORIZAŢIEI DE PUNERE PE PIAŢÃ
AstraZeneca AB
SE-151 85 Södertälje
Suedia
8. NUMĂRUL(ELE) AUTORIZAŢIEI DE PUNERE PE PIAŢĂ
EU/1/20/1479/003
EU/1/20/1479/004
9. DATA PRIMEI AUTORIZĂRI SAU A REÎNNOIRII AUTORIZAŢIEI
Data primei autorizări: 05 noiembrie 2020
Data ultimei reînnoiri a autorizației:
10. DATA REVIZUIRII TEXTULUI
Informaţii detaliate privind acest medicament sunt disponibile pe site-ul Agenţiei Europene pentru Medicamente http://www.ema.europa.eu.