EMEND 125mg + 80mg
Rezumatul caracteristicilor produsului (RCP)
- 1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI
- 2. COMPOZIŢIA CALITATIVĂ ŞI CANTITATIVĂ
- 3. FORMA FARMACEUTICĂ
-
4. DATE CLINICE
- 4.1 Indicaţii terapeutice
- 4.2 Doze şi mod de administrare
- 4.3 Contraindicaţii
- 4.4 Atenţionări şi precauţii speciale pentru utilizare
- 4.5 Interacţiuni cu alte medicamente şi alte forme de interacţiune
- 4.6 Fertilitatea, sarcina şi alăptarea
- 4.7 Efecte asupra capacităţii de a conduce vehicule şi de a folosi utilaje
- 4.8 Reacţii adverse
- 4.9 Supradozaj
- 5. PROPRIETĂŢI FARMACOLOGICE
- 6. PROPRIETĂŢI FARMACEUTICE
- 7. DEŢINĂTORUL AUTORIZAŢIEI DE PUNERE PE PIAŢĂ
- 8. NUMĂRUL(ELE) AUTORIZAŢIEI DE PUNERE PE PIAŢĂ
- 9. DATA PRIMEI AUTORIZĂRI SAU A REÎNNOIRII AUTORIZAŢIEI
- 10. DATA REVIZUIRII TEXTULUI
- 1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI
- 2. COMPOZIŢIA CALITATIVĂ ŞI CANTITATIVĂ
- 3. FORMA FARMACEUTICĂ
-
4. DATE CLINICE
- 4.1 Indicaţii terapeutice
- 4.2 Doze şi mod de administrare
- 4.3 Contraindicaţii
- 4.4 Atenţionări şi precauţii speciale pentru utilizare
- 4.5 Interacţiuni cu alte medicamente şi alte forme de interacţiune
- 4.6 Fertilitatea, sarcina şi alăptarea
- 4.7 Efecte asupra capacităţii de a conduce vehicule şi de a folosi utilaje
- 4.8 Reacţii adverse
- 4.9 Supradozaj
- 5. PROPRIETĂŢI FARMACOLOGICE
- 6. PROPRIETĂŢI FARMACEUTICE
- 7. DEŢINĂTORUL AUTORIZAŢIEI DE PUNERE PE PIAŢĂ
- 8. NUMĂRUL(ELE) AUTORIZAŢIEI DE PUNERE PE PIAŢĂ
- 9. DATA PRIMEI AUTORIZĂRI SAU A REÎNNOIRII AUTORIZAŢIEI
- 10. DATA REVIZUIRII TEXTULUI
1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI
EMEND 125 mg capsule
EMEND 80 mg capsule
2. COMPOZIŢIA CALITATIVĂ ŞI CANTITATIVĂ
Fiecare capsulă de 125 mg conţine aprepitant 125 mg. Fiecare capsulă de 80 mg conţine aprepitant 80 mg.
Excipient cu efect cunoscut
Fiecare capsulă conţine zahăr 125 mg (în capsula de 125 mg).
Excipient cu efect cunoscut
Fiecare capsulă conţine zahăr 80 mg (în capsula de 80 mg).
Pentru lista tuturor excipienţilor, vezi pct. 6.1.
3. FORMA FARMACEUTICĂ
Capsule.
Capsulele de 125 mg sunt opace, cu corp de culoare albă şi capac de culoare roz şi inscripţionate radial, pe corpul capsulei, cu cerneală de culoare neagră, cu „462” şi „125 mg”. Capsulele de 80 mg sunt opace, cu corp şi capac de culoare albă şi inscripţionate radial pe corpul capsulei, cu cerneală de culoare neagră, cu „461” şi „80 mg”.
4. DATE CLINICE
4.1 Indicaţii terapeutice
Prevenirea senzației de greață și vărsăturilor asociate chimioterapiei anticanceroase cu potențial emetogen moderat sau mare, la adulți și adolescenți începând cu vârsta de 12 ani.
EMEND 125 mg/80 mg se administrează în cadrul terapiei combinate (vezi pct. 4.2).
4.2 Doze şi mod de administrare
Doze
Adulți
EMEND se administrează timp de 3 zile, în cadrul unei scheme terapeutice care cuprinde un corticosteroid şi un antagonist al receptorilor 5-HT3. Doza recomandată este de 125 mg administrată pe cale orală o dată pe zi, cu o oră înainte de începerea chimioterapiei în ziua 1 şi de 80 mg oral, o dată pe zi, dimineața, în zilele 2 şi 3.
Următoarele scheme terapeutice sunt recomandate adulților pentru prevenirea greţurilor şi vărsăturilor asociate chimioterapiei anticanceroase cu potenţial emetogen:
Regimuri chimioterapice cu potenţial emetogen mare
| Ziua 1 | Ziua 2 | Ziua 3 | Ziua 4 | |
| EMEND | 125 mg oral | 80 mg oral | 80 mg oral | fără |
| Dexametazonă | 12 mg oral | 8 mg oral | 8 mg oral | 8 mg oral |
| Antagonişti ai receptorilor 5-HT3 | Doza standard de antagonişti ai receptorilor 5-HT3. A se vedea informaţiile despre medicament pentru antagonistul receptorilor 5-HT3 selectat, pentru informaţii corespunzătoare privind dozarea | fără | fără | fără |
Dexametazona trebuie administrată cu 30 minute înainte de chimioterapie în ziua 1 şi dimineaţa în zilele 2-4. Doza de dexametazonă este responsabilă de interacţiunile cu substanţa activă.
Regimuri chimioterapice cu potenţial emetogen moderat
| Ziua 1 | Ziua 2 | Ziua 3 | |
| EMEND | 125 mg oral | 80 mg oral | 80 mg oral |
| Dexametazonă | 12 mg oral | fără | fără |
| Antagonişti ai receptorilor 5-HT3 | Doza standard de antagonişti ai receptorilor 5-HT3. A se vedea informaţiile despre medicament pentru antagonistul receptorilor 5-HT3 selectat, pentru informaţii corespunzătoare privind dozarea | fără | fără |
Dexametazona trebuie administrată în ziua 1, cu 30 minute înainte de chimioterapie. Doza de dexametazonă este responsabilă de interacţiunile cu substanţa activă.
Copii și adolescenți
Adolescenți (cu vârsta cuprinsă între 12 și 17 ani)
EMEND se administrează timp de 3 zile, în cadrul unei scheme terapeutice care cuprinde un antagonist al receptorilor 5-HT3. Doza recomandată de EMEND capsule este de 125 mg administrată pe cale orală în ziua 1 și de 80 mg oral, în zilele 2 și 3. EMEND se administrează pe cale orală cu 1 oră înainte de administrarea chimioterapiei în zilele 1, 2 și 3. Dacă nu se administrează chimioterapie în zilele 2 și 3, EMEND trebuie administrat dimineața. Pentru informații corespunzătoare privind dozarea, vezi Rezumatul Caracteristicilor Produsului (RCP) pentru antagonistul receptorilor 5-HT3 selectat. Dacă un corticosteroid, cum este dexametazona, se administrează concomitent cu EMEND, doza de corticosteroid administrată trebuie să fie 50% din doza uzuală (vezi pct. 4.5 și 5.1).
Siguranța și eficacitatea capsulelor de 80 mg și 125 mg nu au fost demonstrate la copii cu vârsta mai mică de 12 ani. Nu sunt disponibile date. Consultați RCP-ul pentru pulbere pentru suspensie orală pentru dozarea corespunzătoare la sugari, copii mici și copii cu vârsta cuprinsă între 6 luni și 12 ani.
Generalități
Datele referitoare la eficacitate în asociere cu alţi corticosteroizi şi antagonişti ai receptorilor 5-HT3 sunt limitate. Pentru informaţii suplimentare referitoare la administrarea în asociere cu corticosteroizi (vezi pct. 4.5). Vă rugăm să consultaţi RCP-ul pentru medicamentele antagonişti ai receptorilor 5-HT3 administrate în asociere.
Grupe speciale de pacienţi
Vârstnici (≥ 65 ani)
Nu este necesară ajustarea dozei la vârstnici (vezi pct. 5.2).
Sex
Nu este necesară ajustarea dozei în funcţie de sex (vezi pct. 5.2).
Insuficienţă renală
Nu este necesară ajustarea dozei la pacienţii cu insuficienţă renală sau la pacienţii cu afecţiune renală în stadiu terminal trataţi cu hemodializă (vezi pct. 5.2).
Insuficienţă hepatică
Nu este necesară ajustarea dozei la pacienţii cu insuficienţă hepatică uşoară. Datele referitoare la pacienţii cu insuficienţă hepatică moderată sunt limitate, iar pentru pacienţii cu insuficienţă hepatică severă nu există date. Aprepitantul trebuie utilizat cu precauţie la aceşti pacienţi (vezi pct. 4.4 şi 5.2).
Mod de administrare
Capsula trebuie înghiţită întreagă.
EMEND poate fi administrat cu sau fără alimente.
4.3 Contraindicaţii
Hipersensibilitate la substanţa activă sau la oricare dintre excipienţii enumeraţi la pct. 6.1.
Administratrea în asociere cu pimozidă, terfenadină, astemizol sau cisapridă (vezi pct. 4.5).
4.4 Atenţionări şi precauţii speciale pentru utilizare
Pacienţi cu insuficienţă hepatică moderată până la severă
Există informaţii limitate referitoare la pacienţii cu insuficienţă hepatică moderată, iar pentru pacienţii cu insuficienţă hepatică severă nu există date. EMEND trebuie utilizat cu precauţie la aceşti pacienţi (vezi pct. 5.2).
Interacţiuni CYP3A4
EMEND trebuie utilizat cu precauţie la pacienţii care primesc concomitent substanţe active administrate oral care sunt metabolizate în principal pe calea CYP3A4 şi au un indice terapeutic mic, cum ar fi ciclosporină, tacrolimus, sirolimus, everolimus, alfentanil, derivați ai alcaloizilor din ergot, fentanil şi chinidină (vezi pct. 4.5). În plus, sunt necesare precauţii speciale la administrarea concomitentă cu irinotecan, deoarece asocierea poate determina o toxicitate crescută.
Administrarea în asociere cu warfarină (un substrat CYP2C9)
La pacienţii aflaţi în cursul unui tratament cronic cu warfarină, este necesară monitorizarea atentă a Raportului Internaţional Normalizat (INR) în timpul tratamentului cu EMEND şi timp de 14 zile după fiecare cură de 3 zile cu EMEND (vezi pct. 4.5).
Administrarea în asociere cu contraceptive hormonale
Eficacitatea contraceptivelor hormonale poate fi redusă pe durata administrării EMEND şi timp de 28 zile după aceea. Pe durata tratamentului cu EMEND şi timp de 2 luni după administrarea ultimei doze trebuie utilizate metode contraceptive alternative non hormonale suplimentare (vezi pct. 4.5).
Excipienţi
EMEND capsule conţine zahăr. Pacienţii cu afecţiuni ereditare rare de tip intoleranţă la fructoză, sindrom de malabsorbţie la glucoză-galactoză sau insuficienţă a zaharazei-izomaltazei nu trebuie să utilizeze acest medicament.
Sodiu
Acest medicament conține sodiu mai puțin de 1 mmol (23 mg) per capsulă, adică practic „nu conține sodiu”.
4.5 Interacţiuni cu alte medicamente şi alte forme de interacţiune
Aprepitant (125 mg/80 mg) este un substrat, un inhibitor moderat şi un inductor al CYP3A4. De asemenea, aprepitant este un inductor al CYP2C9. În timpul tratamentului cu EMEND este inhibată activitatea CYP3A4. După terminarea tratamentului, EMEND determină o inducţie uşoară şi tranzitorie a CYP2C9, a CYP3A4 şi a glucuronoconjugării. Se pare că aprepitantul nu interacţionează cu transportorul glicoproteinei P, fapt sugerat de absenţa interacţiunii aprepitantului cu digoxina.
Efectul aprepitantului asupra farmacocineticii altor substanţe active
Inhibarea CYP3A4
Fiind un inhibitor moderat al activităţii CYP3A4, aprepitant (125 mg/80 mg) poate creşte concentraţiile plasmatice ale substanţelor active metabolizate pe calea CYP3A4, administrate concomitent. În timpul tratamentului timp de 3 zile cu EMEND, expunerea totală pentru substraturile CYP3A4 administrate oral poate creşte de aproximativ 3 ori; este de aşteptat ca efectul aprepitantului asupra concentraţiilor plasmatice ale substraturilor CYP3A4 administrate intravenos să fie mai mic. EMEND nu trebuie administrat concomitent cu pimozidă, terfenadină, astemizol sau cisapridă (vezi pct. 4.3). Inhibarea CYP3A4 de către aprepitant poate determina creşterea concentraţiilor plasmatice ale acestor substanţe active, cu posibila apariţie a unor reacţii adverse grave sau care pot pune viaţa în pericol. Administrarea EMEND concomitent cu substanţe active administrate oral care sunt metabolizate în principal pe calea CYP3A4 şi au un indice terapeutic mic, cum ar fi ciclosporină, tacrolimus, sirolimus, everolimus, alfentanil, diergotamină, ergotamină, fentanil şi chinidină trebuie făcută cu precauţie (vezi pct. 4.4).
Corticosteroizi
Dexametazonă: În cazul administrării concomitente cu EMEND 125 mg/80 mg, doza orală uzuală de dexametazonă trebuie redusă cu aproximativ 50 %. În studii clinice pentru chimioterapia inducătoare de greață şi vărsături (GVIC), alegerea dozei de dexametazonă a fost făcută în funcţie de interacţiunile cu alte substanţe active (vezi pct. 4.2). Administrarea EMEND în ziua 1 în doză de 125 mg în asociere cu 20 mg dexametazonă pe cale orală, iar în zilele 2-5 în doză de 80 mg/zi în asociere cu 8 mg dexametazonă pe cale orală, a crescut ASC pentru dexametazonă, un substrat al CYP3A4, de 2,2 ori în zilele 1 şi 5.
Metilprednisolonă: În cazul asocierii cu EMEND 125 mg/80 mg, doza uzuală de metilprednisolonă administrată pe cale intravenoasă trebuie redusă cu aproximativ 25 %, iar doza uzuală de metilprednisolonă administrată pe cale orală trebuie redusă cu aproximativ 50 %. EMEND, administrat în doză de 125 mg în ziua 1 şi de 80 mg/zi în zilele 2 şi 3, a crescut ASC pentru metilprednisolonă, un substrat pentru CYP3A4, de 1,3 ori în ziua 1 şi de 2,5 ori în ziua 3, atunci când aceasta a fost administrată concomitent, pe cale intravenoasă în doză de 125 mg în ziua 1 şi pe cale orală în doză de 40 mg în zilele 2 şi 3.
În timpul tratamentului continuu cu metilprednisolonă, ASC pentru metilprednisolonă poate scădea mai târziu, în intervalul de 2 săptămâni după iniţierea administrării EMEND, datorită efectului inductor al aprepitantului asupra CYP3A4. Este de aşteptat ca acest efect să fie mai pronunţat în cazul administrării orale a metilprednisolonei.
Medicamente chimioterapice
În studii farmacocinetice, când a fost administrat în regim de 125 mg în ziua 1 şi 80 mg în zilele 2 şi 3, EMEND nu a influenţat farmacocinetica docetaxel administrat intravenos în ziua 1 sau a vinorelbin administrat intravenos în ziua 1 sau ziua 8. Deoarece efectul EMEND pe farmacocinetica administrării orale a substratului CYP3A4 este mai mare decât efectul EMEND pe farmacocinetica administrării intravenoase a substratului CYP3A4, interacţiunea cu medicamentele chimioterapice administrate oral, metabolizate în principal sau parţial de către CYP3A4 (de exemplu, etopozid, vinorelbin) nu poate fi exclusă. La pacienţii cărora li se administrează medicamente metabolizate în principal sau parţial de către CYP3A4 se recomandă precauţie şi monitorizare suplimentară (vezi pct. 4.4). După punerea pe piaţă, în timpul administrării concomitente de aprepitant cu ifosfamidă, au fost raportate evenimente de neurotoxicitate, o posibilă reacţie adversă a ifosfamidei.
Imunosupresoare
Pe durata regimului GVIC de 3 zile, este de aşteptat o creştere tranzitorie moderată urmată de o scădere uşoară a expunerii imunosupresoarelor metabolizate pe calea CYP3A4 (de exemplu, ciclosporină, tacrolimus, everolimus şi sirolimus). Datorită duratei scurte a regimului de 3 zile şi schimbărilor limitate, dependente de timp, privind expunerea, nu este recomandată reducerea dozei de imunosupresor pe durata celor 3 zile de administrare în asociere cu EMEND.
Midazolam
În cazul administrării concomitente a EMEND (125 mg/80 mg) cu midazolam sau alte benzodiazepine metabolizate pe calea CYP3A4 (alprazolam, triazolam), trebuie avute în vedere efectele posibilei creşteri a concentraţiilor plasmatice ale acestor medicamente.
EMEND a crescut ASC pentru midazolam, un substrat sensibil al CYP3A4, de 2,3 ori în ziua 1 şi de 3,3 ori în ziua 5, în cazul în care au fost administrate doze orale unice de 2 mg midazolam în zilele 1 şi 5 ale unei scheme terapeutice cu 125 mg EMEND în ziua 1 şi 80 mg/zi în zilele 2-5.
În alt studiu care a utilizat administrarea intravenoasă de midazolam, EMEND a fost administrat în doză de 125 mg în ziua 1 şi de 80 mg/zi în zilele 2 şi 3, iar midazolam a fost administrat intravenos în doză de 2 mg anterior regimului de 3 zile cu EMEND şi în zilele 4, 8 şi 15. EMEND a crescut ASC pentru midazolam cu 25 % în ziua 4 şi a scăzut ASC pentru midazolam cu 19 % în ziua 8 şi cu 4 % în ziua 15. Aceste efecte nu au fost considerate importante din punct de vedere clinic.
Într-un al treilea studiu în care s-a administrat midazolam intravenos şi oral, EMEND a fost administrat în doză de 125 mg în ziua 1 şi de 80 mg/zi în zilele 2 şi 3, în asociere cu ondansetron 32 mg în ziua 1, dexametazonă 12 mg în ziua 1 şi 8 mg în zilele 2-4. Această asociere (EMEND, ondansetron şi dexametazonă) a scăzut ASC pentru midazolam administrat oral cu 16 % în ziua 6, cu 9 % în ziua 8, cu 7 % în ziua 15 şi cu 17% în ziua 22. Aceste efecte nu au fost considerate importante din punct de vedere clinic.
S-a efectuat un studiu adiţional privind administrarea intravenoasă de midazolam şi EMEND. Midazolam intravenos 2 mg a fost administrat la 1 oră după administrarea orală a unei doze unice de EMEND de 125 mg. ASC plasmatică a midazolamului a crescut de 1,5 ori. Acest efect nu a fost considerat important din punct de vedere clinic.
Inducţie
Fiind un inductor slab al CYP2C9, al CYP3A4 şi al glucuronoconjugării, aprepitantul poate reduce concentraţiile plasmatice ale substraturilor eliminate pe aceste căi timp de două săptămâni după iniţierea tratamentului. Acest efect poate deveni evident doar după terminarea tratamentului de 3 zile cu EMEND. Inducţia este tranzitorie în cazul substraturilor pentru CYP2C9 şi CYP3A4, iar efectul maxim este atins după 3-5 zile de la terminarea curei de 3 zile cu EMEND. Efectul se menţine câteva zile, ulterior se reduce progresiv şi devine clinic nesemnificativ după două săptămâni de la terminarea tratamentului cu EMEND. Inducţia uşoară a glucuronoconjugării este observată şi după administrarea orală a unei doze de 80 mg de aprepitant timp de 7 zile. Nu sunt disponibile date referitoare la efectele asupra CYP2C8 şi CYP2C19. Se recomandă precauţie la administrarea în această perioadă de timp a warfarinei, acenocumarolului, tolbutamidei, fenitoinei sau a altor substanţe active cunoscute a fi metabolizate pe calea CYP2C9.
Warfarină
La pacienţii aflaţi în tratament cronic cu warfarină, timpul de protrombină (INR) trebuie monitorizat cu atenţie în timpul tratamentului cu EMEND şi timp de 2 săptămâni după fiecare cură de 3 zile cu EMEND (vezi pct. 4.4). Când a fost administrată o doză unică de 125 mg EMEND în ziua 1 şi de 80 mg/zi în zilele 2 şi 3 la subiecţi sănătoşi, stabilizaţi pe o schemă de tratament cronic cu warfarină, nu s-a observat niciun efect al EMEND asupra ASC plasmatice a warfarinei R(+) sau S(-) determinată în ziua 3; cu toate acestea, s-a observat o reducere cu 34 % a concentraţiei minime de warfarină S(-) (substrat al CYP2C9) însoţită de o reducere cu 14 % a INR la 5 zile după terminarea tratamentului cu EMEND.
Tolbutamidă
În cazul administrării orale a unor doze unice de 500 mg tolbutamidă anterior regimului de 3 zile de EMEND şi în zilele 4, 8 şi 15, EMEND, administrat în doză de 125 mg în ziua 1 şi de 80 mg/zi în zilele 2 şi 3, a scăzut ASC pentru tolbutamidă (substrat al CYP2C9) cu 23 % în ziua 4, cu 28 % în ziua 8 şi cu 15 % în ziua 15.
Contraceptivele hormonale
Eficacitatea contraceptivelor hormonale poate fi redusă în timpul administrării EMEND şi timp de 28 zile după aceea. Pe durata tratamentului cu EMEND şi timp de 2 luni după administrarea ultimei doze, trebuie utilizate metode contraceptive alternative non hormonale suplimentare.
Într-un studiu clinic au fost administrate doze unice dintr-un contraceptiv oral conţinând etinilestradiol şi noretindronă în zilele 1-21 împreună cu EMEND 125 mg în ziua 8 şi 80 mg/zi în zilele 9 şi 10, ondansetron 32 mg intravenos în ziua 8 şi dexametazonă administrată oral în doze de 12 mg în ziua 8 şi 8 mg/zi în zilele 9, 10 şi 11. În zilele 9-21 ale acestui studiu, s-au înregistrat reduceri de până la 64 % ale concentraţiilor bazale de etinilestradiol şi reduceri de până la 60 % ale concentraţiilor bazale de noretindronă.
Antagonişti 5-HT3
În studiile clinice privind interacţiunea, aprepitant nu a prezentat efecte importante din punct de vedere clinic asupra farmacocineticii pentru ondansetron, granisetron sau hidrodolasetron (metabolitul activ al dolasetronului).
Efectul altor medicamente asupra farmacocineticii aprepitantului
Administrarea concomitentă de EMEND cu substanţe active care inhibă activitatea CYP3A4 (de exemplu, ketoconazol, itraconazol, voriconazol, posaconazol, claritromicină, telitromicină, nefazodonă şi inhibitori de protează) trebuie efectuată cu prudenţă, deoarece este de aşteptat ca asocierea să determine creşterea de câteva ori a concentraţiilor plasmatice de aprepitant (vezi pct. 4.4).
Trebuie evitată administrarea concomitentă a EMEND cu substanţe active puternic inductoare ale activităţii CYP3A4 (de exemplu, rifampicină, fenitoină, carbamazepină, fenobarbital), deoarece asocierea determină reducerea concentraţiilor plasmatice de aprepitant, cu posibila reducere a eficacităţii EMEND. Nu se recomandă administrarea concomitentă de EMEND cu preparate din plante medicinale ce conţin sunătoare (Hypericum perforatum).
Ketoconazol
La administrarea unei doze unice de 125 mg aprepitant în ziua 5 a unui regim terapeutic de 10 zile cu ketoconazol în doză de 400 mg/zi, inhibitor puternic al CYP3A4, ASC pentru aprepitant a crescut de aproximativ 5 ori şi timpul mediu terminal de înjumătăţire al aprepitantului a crescut de aproximativ 3 ori.
Rifampicină
La administrarea unei doze unice de 375 mg aprepitant în ziua 9 a unui regim terapeutic de 14 zile cu rifampicină în doză de 600 mg/zi, inductor puternic al CYP3A4, ASC pentru aprepitant a scăzut cu 91 % şi timpul mediu terminal de înjumătăţire a scăzut cu 68 %.
Copii și adolescenți
Au fost efectuate studii privind interacțiunile numai la adulți.
4.6 Fertilitatea, sarcina şi alăptarea
Contracepţia la bărbaţi şi femei
Eficacitatea contraceptivelor hormonale poate fi redusă pe perioada administrării şi timp de 28 zile după administrarea EMEND. Trebuie folosite metode contraceptive alternative non hormonale de rezervă în timpul tratamentului cu EMEND şi timp de 2 luni după ultima doză de EMEND (vezi pct. 4.4 şi 4.5).
Sarcina
Nu sunt disponibile date clinice privind expunerea sarcinilor la aprepitant. Nu s-a stabilit complet potenţialul toxicităţii reproductive a aprepitantului, deoarece în studiile la animale nu au putut fi atinse nivele de expunere superioare celor ale expunerii umane la doza de 125 mg/80 mg. Aceste studii nu au evidenţiat efecte nocive directe sau indirecte asupra sarcinii, dezvoltării embrionare/fetale, naşterii sau dezvoltării postnatale (vezi pct. 5.3). Nu se cunosc efectele potenţiale asupra reproducerii ale modificărilor în reglarea neurokininei. EMEND nu trebuie administrat în timpul sarcinii decât dacă este absolut necesar.
Alăptarea
Aprepitant este excretat în laptele femelelor de şobolani. Nu se cunoaşte dacă aprepitant este excretat în laptele uman; de aceea, alăptarea nu este recomandată în timpul tratamentului cu EMEND.
Fertilitatea
Nu s-a stabilit în mod cert potenţialul aprepitantului de a afecta fertilitatea, deoarece în studiile la animale nu au putut fi atinse nivele de expunere superioare celor ale expunerii umane la doza terapeutică. Aceste studii de fertilitate nu au evidenţiat efecte nocive directe sau indirecte asupra capacităţii de reproducere, fertilităţii, dezvoltării embrionare/fetale sau numărului şi motilităţii spermatozoizilor (vezi pct. 5.3).
4.7 Efecte asupra capacităţii de a conduce vehicule şi de a folosi utilaje
EMEND poate avea o influenţă minoră asupra capacităţii de a conduce vehicule, de a merge cu bicicleta şi de a folosi utilaje. După administrarea EMEND pot să apară ameţeală şi fatigabilitate (vezi pct. 4.8).
4.8 Reacţii adverse
Rezumatul profilului de siguranţă
Profilul de siguranţă al aprepitantului a fost evaluat la aproximativ 6500 adulți în mai mult de 50 studii și 184 copii și adolescenți în 2 studii clinice pediatrice pivot.
Cele mai frecvente reacţii adverse raportate cu o incidenţă mai mare la adulții trataţi cu o schemă terapeutică pe bază de aprepitant, comparativ cu cei cărora li s-a administrat terapie standard, în cursul Chimioterapiei cu Potenţial Emetogen Crescut (CPEC), au fost: sughiţ (4,6 % versus 2,9 %), creşterea alanin aminotransferazei (ALAT) (2,8 % versus 1,1 %), dispepsie (2,6 % versus 2,0 %), constipaţie (2,4 % versus 2,0 %), cefalee (2,0 % versus 1,8 %) şi scăderea apetitului alimentar (2,0 % versus 0,5 %). Cea mai frecventă reacţie adversă raportată cu o incidenţă mai mare la pacienţii trataţi cu o schemă terapeutică pe bază de aprepitant, comparativ cu cei care cărora li s-a administrat tratament standard, în cursul Chimioterapiei cu Potenţial Emetogen Moderat (CPEM) a fost fatigabilitatea (1,4 % versus 0,9 %).
În timpul administrării chimioterapiei anticanceroase cu potențial emetogen, cele mai frecvente reacții adverse raportate cu o incidență mai mare la copii și adolescenți tratați urmând o schemă terapeutică pe bază de aprepitant comparativ cu schema terapeutică de control au fost sughiț (3,3 % comparativ cu 0,0 %) și hiperemie (1,1 % comparativ cu 0,0 %).
Lista reacţiilor adverse în format tabelar
Următoarele reacţii adverse au fost observate într-o analiză cumulată a studiilor CPEC şi CPEM cu o incidenţă mai mare la aprepitant decât la terapia standard la adulți sau copii și adolescenți sau în utilizarea după punerea pe piaţă. Categoriile de frecvență din tabel se bazează pe studiile la adulți; frecvențele observate în studiile la copii și adolescenți au fost similare sau mai scăzute, cu excepția cazurilor prezentate în tabel. Unele RA mai puțin frecvente la populația adultă nu au fost observate în studiile la copii și adolescenți.
Frecvenţele sunt definite astfel: foarte frecvente (≥ 1/10); frecvente (≥ 1/100 şi < 1/10); mai puţin frecvente (≥ 1/1000 şi < 1/100); rare (≥ 1/10000 şi < 1/1000) şi foarte rare (< 1/10000), cu frecvenţă necunoscută (care nu poate fi estimată din datele disponibile).
| Clasificare pe aparate, sisteme şi organe | Reacţia adversă | Frecvenţa |
|---|---|---|
| Infecţii şi infestări | ||
| candidoză, infecţie stafilococică | rare | |
| Tulburări hematologice şi limfatice | neutropenie febrilă, anemie | mai puţin frecvente |
| Tulburări ale sistemului imunitar | reacţii de hipersensibilitate incluzând reacţii anafilactice | necunoscută |
| Tulburări metabolice şi de nutriţie | scăderea apetitului alimentar | frecvente |
| polidipsie | rare | |
| Tulburări psihice | anxietate | mai puţin frecvente |
| dezorientare, dispoziţie euforică | rare | |
| Tulburări ale sistemului nervos | cefalee | frecvente |
| ameţeală, somnolenţă | mai puţin frecvente | |
| tulburare cognitivă, letargie, disgeuzie | rare | |
| Tulburări oculare | ||
| conjunctivită | rare | |
| Tulburări acustice şi vestibulare | tinitus | rare |
| Tulburări cardiace | palpitaţii | mai puţin frecvente |
| bradicardie, tulburări cardiovasculare | rare | |
| Tulburări vasculare | ||
| bufeuri/hiperemie | mai puţin frecvente | |
| Tulburări respiratorii, toracice şi mediastinale | sughiţ | frecvente |
| durere orofaringiană, strănut, tuse, creşterea secreţiilor nazale, iritaţie faringiană | rare | |
| Tulburări gastro-intestinale | constipaţie, dispepsie | frecvente |
| eructaţie, greaţă†, vărsături†, boală de reflux gastro-esofagian, durere abdominală, xerostomie, flatulenţă | mai puţin frecvente | |
| ulcer duodenal perforat, stomatită, distensie abdominală, materii fecale tari, colită neutropenică | rare | |
| Tulburări cutanate şi ale ţesutului subcutanat | erupţii cutanate tranzitorii, acnee | mai puţin frecvente |
| reacţie de fotosensibilitate, hiperhidroză, seboree, leziuni cutanate, erupţii cutanate tranzitorii pruriginoase, sindrom Stevens-Johnson/necroliză epidermică toxică | rare | |
| prurit, urticarie | necunoscută | |
| Tulburări musculo-scheletice şi ale ţesutului conjunctiv | slăbiciune musculară, spasme musculare | rare |
| Tulburări renale şi ale căilor urinare | disurie | mai puţin frecvente |
| polakiurie | rare | |
| Tulburări generale şi la nivelul locului de administrare | fatigabilitate | frecvente |
| astenie, stare de rău general | mai puţin frecvente | |
| edeme, disconfort toracic, tulburări de mers | rare | |
| Investigaţii diagnostice | creşterea ALAT | frecvente |
| creşterea ASAT, creşterea fosfatazei alcaline | mai puţin frecvente | |
| hematurie, hiponatremie, scădere ponderală, scăderea numărului de neutrofile, glucozurie, eliminare crescută de urină | rare | |
†Greaţa şi vărsăturile au reprezentat parametrii de eficacitate în primele 5 zile după tratamentul chimioterapic şi au fost raportate ca reacţii adverse numai după aceea.
Descrierea reacţiilor adverse selectate
Profilurile reacţiilor adverse la adulți pentru extensia Multiple-Cycle (cicluri multiple) din studiile CPEC şi CPEM de până la 6 cicluri suplimentare de chimioterapie au fost, în general, similare cu cel observat la ciclul 1.
Într-un studiu clinic suplimentar controlat cu comparator activ la 1169 pacienţi adulți cărora li s-a administrat aprepitant şi CPEC, profilul reacţiilor adverse a fost în general similar celui observat în alte studii CPEC cu aprepitant.
Studii non-GVIC
Reacţii adverse suplimentare au fost observate la pacienţii adulți trataţi cu o doză unică de 40 mg de aprepitant pentru greţurile şi vărsăturile postoperatorii (GVPO) cu o incidenţă mai mare decât cu ondansetron: dureri în etajul abdominal superior, sunete intestinale anormale, constipaţie*, disartrie, dispnee, hipoestezie, insomnie, mioză, greaţă, perturbări senzoriale, disconfort stomacal, subileus*, acuitate vizuală redusă, wheezing.
*Raportate la pacienţii care utilizează o doză mai mare de aprepitant.
Raportarea reacţiilor adverse suspectate
Raportarea reacţiilor adverse suspectate după autorizarea medicamentului este importantă. Acest lucru permite monitorizarea continuă a raportului beneficiu/risc al medicamentului. Profesioniştii din domeniul sănătăţii sunt rugaţi să raporteze orice reacţie adversă suspectată prin intermediul sistemului naţional de raportare, astfel cum este menţionat în Anexa V.
4.9 Supradozaj
În cazul unui supradozaj trebuie întreruptă administrarea EMEND şi trebuie efectuate tratament general de susţinere a funcţiilor vitale şi monitorizare. Datorită acţiunii antiemetice a aprepitantului, este posibil ca emeza indusă de un medicament să nu fie eficientă în aceste cazuri.
Aprepitant nu poate fi îndepărtat prin hemodializă.
5. PROPRIETĂŢI FARMACOLOGICE
5.1 Proprietăţi farmacodinamice
Grupa farmacoterapeutică: Antiemetice şi medicamente pentru combaterea greţei, codul ATC: A04AD12
Aprepitant este un antagonist selectiv, cu afinitate înaltă, al receptorilor neurokininici 1 (NK1) pentru substanţa P umană.
Schema terapeutică de 3 zile pe bază de aprepitant la adulți
În 2 studii randomizate, de tip dublu-orb, care au cuprins un număr total de 1094 pacienţi adulți trataţi cu chimioterapie care a inclus cisplatină în doză ≥ 70 mg/m2, administrarea aprepitantului în asociere cu o schemă terapeutică de ondansetron/dexametazonă (vezi pct. 4.2) a fost comparată cu o schemă terapeutică standard (placebo plus ondansetron 32 mg administrat intravenos în ziua 1 plus dexametazonă administrată pe cale orală 20 mg în ziua 1 şi 8 mg de 2 ori pe zi în zilele 2-4). Deşi în studiile clinice a fost folosită doza de 32 mg ondansetron administrat intravenos, aceasta nu mai este doza recomandată. A se vedea informaţiile despre medicament pentru antagonistul receptorilor 5-HT3 selectat pentru informaţii corespunzătoare privind dozarea.
Evaluarea eficacităţii s-a bazat pe următorul obiectiv compus: răspuns complet (definit ca absenţa episoadelor emetice şi absenţa administrării terapiei de salvare), în principal în timpul ciclului 1. Rezultatele au fost evaluate pentru fiecare studiu în mod individual şi pentru cele 2 studii combinate.
În Tabelul 1 este prezentat un rezumat al principalelor rezultate ale analizei combinate ale celor două studii.
Tabelul 1
Procentul răspunsurilor la pacienţii adulți la care s-a administrat chimioterapie cu potenţial emetogen crescut, pe grupe de tratament şi fază — Ciclul 1
Schemă Tratament Diferenţe* terapeutică standard
OBIECTIVE COMPUSE pe bază de (N= 524)† aprepitant % % (IÎ 95%) (N= 521)†
%
| Răspuns complet (fără emeză şi fără terapie de salvare) | ||||
|---|---|---|---|---|
| Global (0-120 ore) | 67,7 | 47,8 | 19,9 | (14,0; 25,8) |
| 0-24 ore | 86,0 | 73,2 | 12,7 | (7,9; 17,6) |
| 25-120 ore | 71,5 | 51,2 | 20,3 | (14,5; 26,1) |
| OBIECTIVE INDIVIDUALE | ||||
| Fără emeză (fără episoade de emeză, indiferent de utilizarea sau nu a terapiei de salvare) | ||||
| Global (0-120 ore) | 71,9 | 49,7 | 22,2 | (16,4; 28,0) |
| 0-24 ore | 86,8 | 74,0 | 12,7 | (8,0; 17,5) |
| 25-120 ore | 76,2 | 53,5 | 22,6 | (17,0; 28,2) |
| Fără greţuri semnificative (VAS maxim < 25 mm pe o scală între 0-100 mm) | ||||
| Global (0-120 ore) | 72,1 | 64,9 | 7,2 | (1,6; 12,8) |
| 25-120 ore | 74,0 | 66,9 | 7,1 | (1,5; 12,6) |
* Intervalele de încredere au fost calculate fără ajustări în funcţie de sex şi chimioterapia concomitentă, care au fost incluse în analiza iniţială a riscului relativ şi a modelelor logistice.
† Un pacient din schema terapeutică pe bază de aprepitant a prezentat date doar în faza acută şi a fost exclus din analizele globale din faza finală; un pacient din schema terapeutică standard a prezentat date doar în faza finală şi a fost exclus din analizele globale din faza acută.
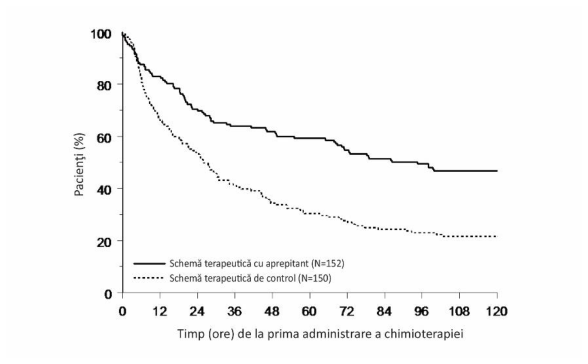
Intervalul estimat până la prima emeză în analiza combinată este ilustrat prin curba Kaplan-Meier din Figura 1.
Figura 1
Procentul de pacienţi adulți trataţi cu chimioterapie cu potenţial emetogen crescut care nu a prezentat emeză pe intervalul de timp – Ciclul 1
100%
Schemă terapeutică cu aprepitant (N=520) Terapie standard (N=523)
90%
Procent de pacienţi
80%
70%
60%
50%
40%
0
0 36 72 108
12 24 48 60 84 96 120
Timp (ore)
Diferenţe semnificative statistic în ceea ce priveşte eficacitatea au fost, de asemenea, observate în fiecare dintre cele 2 studii analizate individual.
În aceleaşi 2 studii clinice, 851 pacienţi adulți au continuat tratamentul în extensia Multiple-Cycle (cicluri multiple) până la 5 cicluri suplimentare de chimioterapie. Aparent, eficacitatea schemei terapeutice pe bază de aprepitant s-a menţinut pe durata tuturor ciclurilor.
Într-un studiu randomizat, dublu-orb, care a cuprins un număr total de 866 pacienţi adulți (864 femei, 2 bărbaţi) trataţi cu chimioterapie care a inclus ciclofosfamidă 750-1500 mg/m2, sau ciclofosfamidă 500-1500 mg/m2 şi doxorubicină (≤ 60 mg/m2) sau epirubicină (≤ 100 mg/m2), administrarea aprepitantului în asociere cu o schemă terapeutică cu ondansetron/dexametazonă (vezi pct. 4.2) a fost comparată cu terapia standard (placebo plus ondansetron 8 mg administrat pe cale orală-de 2 ori pe zi în ziua 1 şi la fiecare 12 ore în zilele 2 şi 3-plus dexametazonă 20 mg administrată pe cale orală în ziua 1).
Evaluarea eficacităţii s-a bazat pe un obiectiv compus: răspuns complet (definit ca absenţa episoadelor emetice şi absenţa administrării terapiei de salvare), în principal în timpul ciclului 1.
Un rezumat al principalelor rezultate ale studiului este prezentat în Tabelul 2.
Tabelul 2
Procentul răspunsurilor la pacienţii adulți pe grupe de tratament şi fază — Ciclul 1 Chimioterapie cu potenţial emetogen moderat
Schemă Tratament Diferenţe* terapeutică standard
OBIECTIVE COMPUSE pe bază de (N= 424) aprepitant % % (IÎ 95%) (N= 433)†
%
| Răspuns complet (fără emeză şi fără terapie de salvare) | ||||
|---|---|---|---|---|
| Global (0-120 ore) 0-24 ore 25-120 ore OBIECTIVE INDIVIDUALE | 50,8 75,7 55,4 | 42,5 69,0 49,1 | 8,3 6,7 6,3 | (1,6; 15,0) (0,7; 12,7) (-0,4; 13,0) |
| Fără emeză (fără episoade de emeză, indiferent de utilizarea sau nu a terapiei | ||||
| de salvare) Global (0-120 ore) 0-24 ore 25-120 ore | 75,7 87,5 80,8 | 58,7 77,3 69,1 | 17,0 10,2 11,7 | (10,8; 23,2) (5,1; 15,3) (5,9; 17,5) |
| Fără greţuri semnificative (VAS maxim < 25 mm pe o scală între 0-100 mm) | ||||
| Global (0-120 ore) | 60,9 | 55,7 | 5,3 | (-1,3; 11,9) |
| 0-24 ore | 79,5 | 78,3 | 1,3 | (-4,2; 6,8) |
| 25-120 ore | 65,3 | 61,5 | 3,9 | (-2,6; 10,3) |
* Intervalele de încredere au fost calculate fără ajustări în funcţie de grupa de vârstă (< 55 ani, ≥ 55 ani) şi de grupul investigator, care au fost incluse în analiza iniţială a riscului relativ şi a modelelor logistice. † Un pacient din schema terapeutică pe bază de aprepitant a prezentat date doar în faza acută şi a fost exclus din analizele globale din faza finală.
În acelaşi studiu clinic, 744 pacienţi adulți au continuat tratamentul în extensia Multiple-Cycle (cicluri multiple) până la 3 cicluri suplimentare de chimioterapie. Aparent, eficacitatea schemei terapeutice pe bază de aprepitant s-a menţinut pe durata tuturor ciclurilor.
Într-un al doilea studiu clinic multicentric, randomizat, dublu-orb, cu grupuri paralele, schema terapeutică pe bază de aprepitant a fost comparată cu schema terapeutică standard la 848 pacienţi adulți (652 femei, 196 bărbaţi) trataţi cu o schemă chimioterapeutică care a inclus oricare doză intravenoasă de oxaliplatin, carboplatin, epirubicină, idarubicină, ifosfamidă, irinotecan, daunorubicină, doxorubicină; ciclofosfamidă intravenoasă (< 1500 mg/m2); sau citarabină intravenoasă (> 1 g/m2). Pacienţilor trataţi cu schema terapeutică pe bază de aprepitant li s-a administrat chimioterapie pentru o varietate de tipuri de tumori incluzând 52 % cu cancer de sân, 21 % cu cancere gastro-intestinale inclusiv cancer colorectal, 13 % cu cancer pulmonar şi 6 % cu cancere ginecologice. Schema terapeutică pe bază de aprepitant în asociere cu o schemă terapeutică cu ondansetron/dexametazonă (vezi pct. 4.2) a fost comparată cu terapia standard (placebo în asociere cu ondansetron 8 mg administrat pe cale orală (de 2 ori pe zi în ziua 1 şi la intervale de 12 ore în zilele 2 şi 3) plus dexametazonă 20 mg administrată pe cale orală în ziua 1).
Evaluarea eficacităţii s-a bazat pe următoarele criterii principale şi secundare finale de evaluare: absența vărsăturilor în perioada globală (0 până la 120 ore post-chimioterapie), evaluarea siguranţei şi tolerabilităţii schemei terapeutice pe bază de aprepitant pentru greața şi vărsăturile induse de chimioterapie (GVIC), şi răspuns complet (definit ca absenţa vărsăturilor şi absenţa administrării terapiei de salvare) în perioada globală (0 până la 120 ore post-chimioterapie). În plus, absenţa greţei semnificative în perioada globală (0 până la 120 ore post-chimioterapie) a fost evaluată ca un criteriu explorator final de evaluare şi în fazele acute şi întârziate ca analiză post-hoc.
Un rezumat al principalelor rezultate ale studiului este prezentat în Tabelul 3.
Tabel 3
Procentul răspunsurilor la pacienţii adulți pe grupe de tratament şi fază pentru Studiul 2 — Ciclul 1 Chimioterapie cu potenţial emetogen moderat
Schemă Tratament Diferenţe * terapeutică standard pe bază de (N= 406) aprepitant % % (IÎ 95 %) (N= 425)
%
| Răspuns complet (fără emeză şi fără terapie de salvare) | ||||
|---|---|---|---|---|
| Global (0-120 ore) | 68,7 | 56,3 | 12,4 | (5,9; 18,9) |
| 0-24 ore | 89,2 | 80,3 | 8,9 | (4,0; 13,8) |
| 25-120 ore | 70,8 | 60,9 | 9,9 | (3,5; 16,3) |
| Fără emeză (fără episoade de emeză, indiferent de utilizarea sau nu a terapiei de salvare) | ||||
| Global (0-120 ore) | 76,2 | 62,1 | 14,1 | (7,9; 20,3) |
| 0-24 ore | 92,0 | 83,7 | 8,3 | (3,9; 12,7) |
| 25-120 ore | 77,9 | 66,8 | 11,1 | (5,1; 17,1) |
| Fără greţuri semnificative (VAS maxim < 25 mm pe o scală între 0-100 mm) | ||||
| Global (0-120 ore) | 73,6 | 66,4 | 7,2 | (1,0; 13,4) |
| 0-24 ore | 90,9 | 86,3 | 4,6 | (0,2, 9,0) |
| 25-120 ore | 74,9 | 69,5 | 5,4 | (-0,7, 11,5) |
* Intervalele de încredere au fost calculate fără ajustări în funcţie de sex şi regiune, care au fost incluse în analiza iniţială utilizând modelele logistice.
Beneficiul schemei terapeutice asociate cu aprepitant la întreaga populaţie participantă la studiu a fost determinat în principal de rezultatele observate la pacienţii cu un control slab obţinut cu schema terapeutică standard, cum ar fi la femei, deşi rezultatele au fost numeric mai bune indiferent de vârstă, tip de tumoră sau sex. A fost obţinut răspuns complet la schema terapeutică pe bază de aprepitant şi respectiv la tratamentul standard la 209/304 (65 %) şi 161/320 (50 %) la femei şi 83/101 (82 %) şi 68/87 (78 %) la bărbaţi.
Copii şi adolescenţi
Într-un studiu clinic randomizat, dublu-orb, controlat cu comparator activ, care a inclus 302 copii și adolescenți (cu vârsta cuprinsă între 6 luni și 17 ani) cărora li s-au administrat chimioterapie cu potențial emetogen moderat sau mare, schema terapeutică pe bază de aprepitant a fost comparată cu o schemă terapeutică de control pentru prevenirea GVIC. Eficacitatea schemei terapeutice cu aprepitant a fost evaluată într-un singur ciclu (Ciclul 1). Pacienții au avut posibilitatea de a urma în mod deschis tratamentul cu aprepitant în cicluri ulterioare (Cicluri opționale 2-6); cu toate acestea, eficacitatea nu a fost evaluată în aceste cicluri opționale. Schema terapeutică pe bază de aprepitant pentru adolescenții cu vârsta cuprinsă între 12 și 17 ani (n=47) a constat în EMEND capsule 125 mg pe cale orală în ziua 1 și 80 mg/zi în ziua 2 și 3, în asociere cu ondansetron în ziua 1. Schema terapeutică pe bază de aprepitant pentru copiii cu vârsta cuprinsă între 6 luni și 12 ani (n=105) a constat în EMEND pulbere pentru suspensie orală 3,0 mg/kg (până la 125 mg) pe cale orală în ziua 1 și 2,0 mg/kg (până la 80 mg) pe cale orală în zilele 2 și 3, în asociere cu ondansetron în ziua 1. Schema terapeutică de control la adolescenții cu vârsta cuprinsă între 12 și 17 ani (n=48) și copiii cu vârsta cuprinsă între 6 luni și 12 ani (n=102) a constat în placebo în loc de aprepitant în zilele 1, 2 și 3 în asociere cu ondansetron în ziua 1. EMEND sau placebo și ondansetron au fost administrate cu 1 oră și respectiv cu 30 minute înainte de inițierea chimioterapiei. Administrarea intravenoasă a dexametazonei a fost permisă în cadrul schemei terapeutice antiemetice pentru copiii și adolescenții din ambele grupe de vârsta, în funcție de decizia medicului. În cazul copiilor și adolescenților care au primit aprepitant, a fost necesară reducerea dozei (50 %) de dexametazonă. Nu a fost necesară reducerea dozei în cazul copiilor și adolescenților care au primit schema terapeutică de control. Dintre copii și adolescenți, la 29 % din cei care au primit schema terapeutică pe bază de aprepitant și la 28 % din cei care au primit schema terapeutică de control, s-a administrat dexametazona ca parte a schemei terapeutice din Ciclul 1.
Activitatea antiemetică a EMEND a fost evaluată pe o perioadă de 5 zile (120 ore) după inițierea chimioterapiei în ziua 1. Obiectivul clinic primar a fost răspunsul complet în faza tardivă (25 până la 120 ore după inițierea chimioterapiei) în Ciclul 1. Un rezumat al principalelor rezultate ale studiului este prezentat în Tabelul 4.
Tabelul 4
Numărul (%) de copii și adolescenți cu răspuns complet și fără vărsături pe grupe de tratament și fază – Ciclul 1 (populație în intenție de tratament)
| Schema terapeutică pe bază de aprepitant n/m (%) | Schema terapeutică de control n/m (%) | |
| OBIECTIV CLINIC PRIMAR | ||
|---|---|---|
| Răspuns complet*–Faza tardivă | 77/152 (50,7)† | 39/150 (26,0) |
| ALTE OBIECTIVE CLINICE PRESPECIFICATE | ||
| Răspuns complet*–Faza acută | 101/152 (66,4)‡ | 78/150 (52,0) |
| Răspuns complet*–Faza globală | 61/152 (40,1)† | 30/150 (20,0) |
| Fără vărsături§–Faza globală | 71/152 (46,7)† | 32/150 (21,3) |
*Răspuns complet = Fără vărsături sau eructație sau reflex de vărsăturăși fără utilizarea medicației de urgență. †p < 0,01 atunci când se compară cu schema terapeutică de control. ‡p < 0,05 atunci când se compară cu schema terapeutică de control. §Fărăvărsături = fără vărsături sau eructație saureflex de vărsătură. n/m = Număr de pacienți cu răspunsul dorit/număr de pacienți incluși în interval. Faza acută: 0 până la 24 ore după inițierea chimioterapiei. Faza tardivă: 25până la 120 ore după inițierea chimioterapiei. Faza globală: 0 până la 120 ore după inițierea chimioterapiei. | ||
Timpul estimat până la primele vărsături după iniţierea tratamentului chimioterapic a fost mai mare în cazul schemei terapeutice pe bază de aprepitant (timpul median estimat până la primele vărsături a fost de 94,5 ore) comparativ cu grupul care a primit schema terapeutică de control (timpul median estimat până la primele vărsături a fost de 26,0 ore) așa cum arată curbele Kaplan-Meier din Figura 2.
Figura 2
Timp până la primul episod de vărsături de la începutul administrării chimioterapiei - copii și adolescenți în faza globală-Ciclul 1 (populație în intenție de tratament)
O analiză a eficacității la nivelul subpopulaţiilor din Ciclul 1 a demonstrat că, indiferent de categoria de vârstă, sex, utilizarea dexametazonei pentru profilaxia antiemetică şi de potenţialul emetogen al chimioterapiei, schema terapeutică pe bază de aprepitant a oferit un control mai bun faţă de cel obţinut cu schema terapeutică de control în ceea ce priveşte obiectivele clinice de răspuns complet.
5.2 Proprietăţi farmacocinetice
Aprepitant prezintă o farmacocinetică nelineară. Atât clearance-ul, cât şi biodisponibilitatea absolută scad odată cu creşterea dozei.
Absorbţie
Biodisponibilitatea absolută medie a aprepitantului după administrare orală este de 67% pentru capsula de 80 mg şi de 59 % pentru capsula de 125 mg. Media concentraţiei plasmatice maxime (Cmax) a aprepitantului s-a înregistrat la aproximativ 4 ore (tmax). Administrarea orală a unei capsule cu un mic dejun standard de aproximativ 800 Kcal a determinat o creştere de până la 40 % a ASC pentru aprepitant. Această creştere nu este considerată semnificativă din punct de vedere clinic.
Farmacocinetica aprepitantului este nelineară în limitele dozelor clinice. La adulţi tineri sănătoşi, creşterea ASC0-∞ a fost cu 26 % mai mare decât raportul dozelor unice de 80 mg şi 125 mg administrate postprandial.
După administrarea pe cale orală a unei doze unice de 125 mg EMEND în ziua 1 şi 80 mg o dată pe zi în zilele 2 şi 3, ASC0-24ore (medie ± DS) a fost de 19,6 ± 2,5 µg•oră/ml şi 21,2 ± 6,3 µg•oră/ml în zilele 1 şi respectiv 3. Cmax a fost de 1,6 ± 0,36 µg/ml şi 1,4 ± 0,22 µg/ml în zilele 1 şi respectiv 3.
Distribuţie
Aprepitantul se leagă în proporţie mare de proteine, media fiind de 97 %. La om, la starea de echilibru, volumul geometric aparent mediu de distribuţie (Vdss) este de aproximativ 66 l.
Metabolizare
Aprepitantul este supus unei metabolizări extensive. După administrarea intravenoasă a unei doze unice de 100 mg fosaprepitant marcat cu [14C], un promedicament pentru aprepitant, la adulţi tineri sănătoşi, aprepitantul este responsabil pentru aproximativ 19% din radioactivitatea plasmatică determinată după 72 ore, ceea ce indică prezenţa substanţială a metaboliţilor în plasmă. În plasma umană au fost identificaţi 12 metaboliţi ai aprepitantului. Metabolizarea aprepitantului se produce, în principal, prin oxidarea inelului morfolinic şi a catenelor sale, metaboliţii rezultaţi având o activitate slabă. Studiile in vitro care au utilizat microzomi hepatici umani indică faptul că aprepitantul este metabolizat în principal de către CYP3A4, cu o contribuţie potenţială minoră a CYP1A2 şi CYP2C19.
Eliminare
Aprepitantul nu se elimină nemetabolizat prin urină. Metaboliţii sunt excretaţi în urină, iar pe calea excreţiei biliare, în fecale. După administrarea intravenoasă la subiecţi sănătoşi a unei doze unice de 100 mg fosaprepitant marcat cu [14C], un promedicament pentru aprepitant, 57 % din radioactivitate a fost detectată în urină, iar 45 % în materiile fecale.
Clearance-ul plasmatic al aprepitantului este dependent de doză, scăzând cu creşterea acesteia şi variind între aproximativ 60 şi 72 ml/minut în intervalul dozelor terapeutice. Timpul de înjumătăţire variază aproximativ de la 9 la 13 ore.
Farmacocinetica la grupe speciale de pacienţi
Vârstnici: După administrarea orală a unei doze unice de 125 mg aprepitant în ziua 1 şi a unei doze de 80 mg o dată pe zi în zilele 2-5, ASC0-24ore pentru aprepitant a fost cu 21 % mai mare în ziua 1 şi cu 36 % mai mare în ziua 5 la vârstnici (≥ 65 ani), comparativ cu adulţii tineri. Cmax a fost cu 10 % mai mare în ziua 1 şi cu 24 % mai mare în ziua 5 la vârstnici, comparativ cu adulţii tineri. Aceste diferenţe nu au fost considerate semnificative din punct de vedere clinic. Nu este necesară ajustarea dozelor de EMEND la pacienţii vârstnici.
Sex: După administrarea orală a unei doze unice de 125 mg aprepitant, Cmax pentru aprepitant este cu 16 % mai mare la femei, comparativ cu bărbaţii. Timpul de înjumătăţire al aprepitantului este cu 25 % mai mic la femei comparativ cu bărbaţii, iar tmax se înregistrează după aproximativ acelaşi interval de timp. Aceste diferenţe nu au fost considerate semnificative din punct de vedere clinic. Nu este necesară ajustarea dozelor de EMEND în funcţie de sex.
Insuficienţă hepatică: Insuficienţa hepatică uşoară (Child-Pugh clasă A) nu influenţează farmacocinetica aprepitantului în mod semnificativ din punct de vedere clinic. Nu este necesară ajustarea dozei la pacienţii cu insuficienţă hepatică uşoară. Din datele existente nu pot fi trase concluzii referitoare la influenţa insuficienţei hepatice moderate (Child-Pugh clasă B) asupra farmacocineticii aprepitantului. Nu există date clinice sau farmacocinetice la pacienţii cu insuficienţă hepatică severă (Child-Pugh clasă C).
Insuficienţă renală: La pacienţi cu insuficienţă renală severă (Cl creatinină < 30 ml/minut) şi la cei cu afecţiuni renale în stadiu terminal (ARST) care necesită hemodializă a fost administrată o doză unică de 240 mg aprepitant.
La pacienţii cu insuficienţă renală severă, ASC0-∞ pentru aprepitantul total (liber şi legat de proteine) a scăzut cu 21 %, iar Cmax a scăzut cu 32 %, comparativ cu subiecţii sănătoşi. La pacienţii cu ARST care fac hemodializă, ASC0-∞ pentru aprepitantul total a scăzut cu 42 %, iar Cmax a scăzut cu 32 %. Datorită scăderii uşoare a legării aprepitantului de proteine la pacienţii cu afecţiuni renale, ASC pentru aprepitantul liber, farmacologic activ, nu a fost influenţată semnificativ la pacienţii cu insuficienţă renală, comparativ cu subiecţii sănătoşi. Hemodializa efectuată la 4 sau 48 ore după administrarea dozei nu a avut efecte semnificative asupra farmacocineticii aprepitantului; mai puţin de 0,2 % din doză a fost recuperată în dializat.
Nu este necesară ajustarea dozei de EMEND la pacienţi cu insuficienţă renală sau la pacienţi cu ARST care fac hemodializă.
Copii și adolescenți: În cadrul schemei terapeutice de 3 zile, dozarea capsulelor de aprepitant (125/80/80 mg) la pacienți adolescenți (cu vârsta cuprinsă între 12 și 17 ani) a determinat o ASC0-24ore peste 17 µg•oră/ml în ziua 1 cu concentrații (Cmin) la sfârșitul zilelor 2 și 3 peste 0,4 µg/ml la majoritatea pacienților. Valoarea mediană a concentrației plasmatice maxime (Cmax) a fost aproximativ 1,3 µg/ml în ziua 1, înregistrându-se la aproximativ 4 ore. În cadrul schemei terapeutice de 3 zile, dozarea pulberii pentru suspensie orală de aprepitant (3/2/2 mg/kg) la pacienți cu vârsta cuprinsă între 6 luni și 12 ani a determinat o ASC0-24ore peste 17 µg•oră/ml în ziua 1 cu concentrații (Cmin) la sfârșitul zilelor 2 și 3 peste 0,1 µg/ml la majoritatea pacienților. Valoarea mediană a concentrației plasmatice maxime (Cmax) a fost aproximativ 1,2 µg/ml în ziua 1, înregistrându-se între 5 și 7 ore.
O analiză farmacocinetică populațională a aprepitantului la copii și adolescenți (cu vârsta cuprinsă între 6 luni și 17 ani) sugerează faptul că sexul și rasa nu au un efect semnificativ din punct de vedere clinic asupra farmacocineticii aprepitantului.
Relaţia între concentraţie şi efect
Utilizând un trasor cu specificitate crescută pentru receptorii NK1, studiile tomografice cu emisie de pozitroni (PET) efectuate la bărbaţi tineri sănătoşi au demonstrat faptul că aprepitantul pătrunde în creier şi se leagă de receptorii NK1 într-un mod dependent de doză şi de concentraţia plasmatică. Concentraţiile plasmatice de aprepitant obţinute după o schemă terapeutică de 3 zile cu EMEND la adulți furnizează o saturare de peste 95 % a receptorilor cerebrali NK1.
5.3 Date preclinice de siguranţă
Datele preclinice nu au evidenţiat niciun risc special pentru om, pe baza studiilor convenţionale de toxicitate după administrare unică şi după doze repetate, genotoxicitate, potenţial carcinogen, de toxicitate asupra funcţiei de reproducere și dezvoltării. Cu toate acestea, trebuie reţinut că expunerea sistemică la rozătoare a fost similară sau chiar inferioară expunerii terapeutice la om la doza de 125 mg/80 mg. În particular, deşi în studiile asupra funcţiei reproductive nu s-au înregistrat reacţii adverse la nivelele de expunere umană, expunerile la animale nu sunt suficiente pentru a evalua în mod adecvat riscurile potenţiale la om.
Într-un studiu privind toxicitatea juvenilă la șobolanii tratați din ziua postnatală 10 până în ziua 63, aprepitantul a condus la o deschidere vaginală timpurie la femele, începând cu doza de 250 mg/kg de două ori pe zi și la o separare prepuţială întârziată la masculi, începând cu doza de 10 mg/kg de două ori pe zi. Nu au existat limite privind expunerea relevantă clinic. Nu s-au înregistrat efecte legate de tratament asupra împerecherii, fertilității sau privind supraviețuirea embrionară/fetală și nu s-au produs modificări patologice la nivelul organelor reproducătoare. Într-un studiu privind toxicitatea juvenilă la câinii tratați din ziua postnatală 14 până la ziua 42, s-a observat reducerea greutății testiculare și a dimensiunii celulelor Leydig la masculi la doza de 6 mg/kg/zi și creșterea greutății uterului, hipertrofia uterului și a colului uterin și edemul țesutului vaginal au fost observate la femele începând cu doza de 4 mg/kg/zi. Nu au existat limite privind expunerea relevantă clinic la aprepitant. În cazul tratamentului pe termen scurt conform schemei terapeutice recomandate ale dozelor, aceste rezultate nu sunt considerate a fi relevante clinic.
6. PROPRIETĂŢI FARMACEUTICE
6.1 Lista excipienţilor
Conţinutul capsulei
Zahăr
Celuloză microcristalină (E 460)
Hidroxipropilceluloză (E 463)
Laurilsulfat de sodiu
Capsula (125 mg)
Gelatină
Dioxid de titan (E 171)
Oxid roşu de fer (E 172)
Oxid galben de fer (E 172)
Capsula (80 mg)
Gelatină
Dioxid de titan (E 171)
Cerneala de inscripţionare
Shellac
Hidroxid de potasiu
Oxid negru de fer (E 172)
6.2 Incompatibilităţi
Nu este cazul.
6.3 Perioada de valabilitate
4 ani
6.4 Precauţii speciale pentru păstrare
A se păstra în ambalajul original pentru a fi protejat de umiditate.
6.5 Natura şi conţinutul ambalajului
Sunt disponibile diferite mărimi de ambalaj, conţinând capsule cu diferite dozaje.
Blister de aluminiu conţinând o capsulă de 80 mg.
Blister de aluminiu conţinând două capsule de 80 mg.
5 blistere de aluminiu fiecare conţinând o capsulă de 80 mg.
Blister de aluminiu conţinând o capsulă de 125 mg.
5 blistere de aluminiu fiecare conţinând o capsulă de 125 mg.
Blister de aluminiu conţinând o capsulă de 125 mg şi două de 80 mg.
Este posibil ca nu toate mărimile de ambalaje să fie comercializate.
6.6 Precauţii speciale pentru eliminarea reziduurilor
Fără cerinţe speciale la eliminare.
7. DEŢINĂTORUL AUTORIZAŢIEI DE PUNERE PE PIAŢĂ
Merck Sharp & Dohme B.V.
Waarderweg 39
2031 BN Haarlem
Olanda
8. NUMĂRUL(ELE) AUTORIZAŢIEI DE PUNERE PE PIAŢĂ
EU/1/03/262/001
EU/1/03/262/002
EU/1/03/262/003
EU/1/03/262/004
EU/1/03/262/005
EU/1/03/262/006
9. DATA PRIMEI AUTORIZĂRI SAU A REÎNNOIRII AUTORIZAŢIEI
Data primei autorizări: 11 noiembrie 2003
Data ultimei reînnoiri a autorizaţiei: 22 septembrie 2008
10. DATA REVIZUIRII TEXTULUI
Informaţii detaliate privind acest medicament sunt disponibile pe site-ul Agenţiei Europene pentru Medicamente http://www.ema.europa.eu.
1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI
EMEND 125 mg pulbere pentru suspensie orală
2. COMPOZIŢIA CALITATIVĂ ŞI CANTITATIVĂ
Fiecare plic conţine aprepitant 125 mg. După reconstituire, 1 ml de suspensie orală conţine aprepitant 25 mg.
Excipient(ţi) cu efect cunoscut
Fiecare plic conţine zahăr aproximativ 125 mg şi lactoză 468,7 mg (sub formă anhidră).
Pentru lista tuturor excipienţilor, vezi pct. 6.1.
3. FORMA FARMACEUTICĂ
Pulbere pentru suspensie orală.
Pulbere de culoare roz până la roz deschis.
4. DATE CLINICE
4.1 Indicaţii terapeutice
Prevenirea senzației de greaţă şi vărsăturilor asociate chimioterapiei anticanceroase cu potențial emetogen moderat sau mare la copii, copii mici şi sugari cu vârsta cuprinsă între 6 luni și 12 ani.
EMEND pulbere pentru suspensie orală se administrează în cadrul terapiei combinate (vezi pct. 4.2).
4.2 Doze şi mod de administrare
Doar profesioniștii din domeniul sănătății trebuie să prepare suspensia orală și să măsoare doza de medicament.
Doze
Copii şi adolescenţi
Sugari, copii mici şi copii (cu vârsta cuprinsă între 6 luni și 12 ani şi nu mai puțin de 6 kg) EMEND se administrează timp de 3 zile în cadrul unei scheme terapeutice care cuprinde un antagonist al receptorilor 5-HT3. Doza recomandată de EMEND pulbere pentru suspensie orală se stabileşte în funcţie de greutatea corporală, așa cum este specificat în tabelul de mai jos. EMEND se administrează pe cale orală, cu o oră înainte de începerea chimioterapiei, în zilele 1, 2 şi 3. Dacă nu se administrează chimioterapie în zilele 2 şi 3, EMEND trebuie administrat dimineaţa. Pentru informaţii corespunzătoare privind dozarea, vezi Rezumatul caracteristicilor produsului (RCP) pentru antagonistul receptorilor 5-HT3 selectat. Dacă un corticosteroid, cum este dexametazona, se administrează concomitent cu EMEND, doza de corticosteroid administrată trebuie să fie 50% din doza uzuală (vezi pct. 4.5 şi 5.1).
Doza de EMEND suspensie orală recomandată la copii şi adolescenţi cu vârsta cuprinsă între 6 luni şi 12 ani
| Ziua 1 | Ziua 2 | Ziua 3 | |
| EMEND suspensie orală 25 mg/ml | 3 mg/kg oral Doza maximă 125 mg | 2 mg/kg oral Doza maximă 80 mg | 2 mg/kg oral Doza maximă 80 mg |
Eficacitatea formulării farmaceutice de 125 mg pulbere pentru suspensie orală nu a fost stabilită la copii cu vârsta de 12 ani și peste. Pentru adolescenţi cu vârsta cuprinsă între 12 și 17 ani, EMEND este disponibil sub formă de capsule care conțin aprepitant 80 mg sau 125 mg.
Siguranţa şi eficacitatea EMEND pulbere pentru suspensie orală la sugari cu vârsta sub 6 luni sau cu o greutate corporală sub 6 kg nu au fost stabilite. Nu sunt disponibile date.
Generalităţi
Datele referitoare la eficacitate în asociere cu alţi corticosteroizi şi antagonişti ai receptorilor 5-HT3 sunt limitate. Pentru informaţii suplimentare referitoare la administrarea în asociere cu corticosteroizi (vezi pct. 4.5). Vă rugăm să consultaţi RCP-ul medicamentelor de tip antagonişti ai receptorilor 5-HT3 administrate în asociere.
Grupe speciale de pacienţi
Sex
Nu este necesară ajustarea dozei în funcţie de sex (vezi pct. 5.2).
Insuficienţă renală
Nu este necesară ajutarea dozei la pacienţii cu insuficienţă renală sau la pacienţii cu afecțiune renală în stadiu terminal trataţi cu hemodializă (vezi pct. 5.2).
Insuficienţă hepatică
Nu este necesară ajustarea dozei la pacienţii cu insuficienţă hepatică uşoară. Datele referitoare la pacienţii cu insuficienţă hepatică moderată sunt limitate, iar pentru pacienţii cu insuficienţă hepatică severă nu există date. Aprepitantul trebuie utilizat cu precauţie la aceşti pacienţi (vezi pct. 4.4 şi 5.2).
Mod de administrare
Suspensia orală poate fi administrată cu sau fără alimente.
Pentru detalii privind pregătirea şi administrarea suspensiei, vezi pct. 6.6.
4.3 Contraindicaţii
Hipersensibilitate la substanţa activă sau la oricare dintre excipienţii enumeraţi la pct. 6.1.
Administrarea în asociere cu pimozidă, terfenadină, astemizol sau cisapridă (vezi pct. 4.5).
4.4 Atenţionări şi precauţii speciale pentru utilizare
Pacienţi cu insuficienţă hepatică moderată până la severă
Există informaţii limitate referitoare la pacienţii cu insuficienţă hepatică moderată, iar pentru pacienţii cu insuficienţă hepatică severă nu există date. EMEND trebuie utilizat cu precauţie la aceşti pacienţi (vezi pct. 5.2).
Interacţiuni CYP3A4
EMEND trebuie utilizat cu precauţie la pacienţii care primesc concomitent substanţe active administrate oral care sunt metabolizate în principal pe calea CYP3A4 şi au un indice terapeutic mic, cum sunt ciclosporină, tacrolimus, sirolimus, everolimus, alfentanil, derivați ai alcaloizilor din ergot, fentanil şi chinidină (vezi pct. 4.5). În plus, sunt necesare precauţii speciale la administrarea concomitentă cu irinotecan deoarece asocierea poate determina o toxicitate crescută.
Administrarea în asociere cu warfarină (un substrat CYP2C9)
La pacienţii aflați în cursul unui tratament cronic cu warfarină, este necesară monitorizarea atentă a Raportului Internaţional Normalizat (INR) în timpul tratamentului cu EMEND şi timp de 14 zile după fiecare cură de 3 zile cu EMEND (vezi pct. 4.5).
Administrarea în asociere cu contraceptive hormonale
Eficacitatea contraceptivelor hormonale poate fi redusă pe durata administrării EMEND şi timp de 28 zile după aceea. Pe durata tratamentului cu EMEND şi timp de 2 luni după administrarea ultimei doze trebuie utilizate metode contraceptive alternative non hormonale suplimentare (vezi pct 4.5).
Excipienţi
EMEND pulbere pentru suspensie orală conţine zahăr şi lactoză. Pacienţii cu afecţiuni ereditare rare de tip intoleranţă la fructoză sau galactoză, sindrom de malabsorbţie la glucoză-galactoză, deficit total de lactază sau insuficienţă a zaharazei-izomaltazei nu trebuie să utilizeze acest medicament.
Sodiu
Acest medicament conține sodiu mai puțin de 1 mmol (23 mg) per plic, adică practic „nu conține sodiu”.
4.5 Interacţiuni cu alte medicamente şi alte forme de interacţiune
Aprepitant (125 mg/80 mg) este un substrat, un inhibitor moderat şi un inductor al CYP3A4. De asemenea, aprepitant este un inductor al CYP2C9. În timpul tratamentului cu EMEND este inhibată activitatea CYP3A4. După terminarea tratamentului, EMEND determină o inducţie uşoară şi tranzitorie a CYP2C9, a CYP3A4 şi a glucuronoconjugării. Se pare că aprepitantul nu interacţionează cu transportorul glicoproteinei P, fapt sugerat de absenţa interacţiunii aprepitantului cu digoxina.
Efectul aprepitantului asupra farmacocineticii altor substanţe active
Inhibarea CYP3A4
Fiind un inhibitor moderat al activităţii CYP3A4, aprepitant (125 mg/80 mg) poate creşte concentraţiile plasmatice ale substanţelor active metabolizate pe calea CYP3A4, administrate concomitent. În timpul tratamentului de 3 zile cu EMEND expunerea totală pentru substraturile CYP3A4 administrate oral poate creşte de aproximativ 3 ori; este de aşteptat ca efectul aprepitantului asupra concentraţiilor plasmatice ale substraturilor CYP3A4 administrate intravenos să fie mai mic. EMEND nu trebuie administrat concomitent cu pimozidă, terfenadină, astemizol sau cisapridă (vezi pct. 4.3). Inhibarea CYP3A4 de către aprepitant poate determina creşterea concentraţiilor plasmatice ale acestor substanţe active, cu posibila apariţie a unor reacţii adverse grave sau care pot pune viaţa în pericol. Administrarea EMEND concomitent cu substanţe active administrate oral care sunt metabolizate în principal pe calea CYP3A4 şi au un indice terapeutic mic, cum sunt ciclosporină, tacrolimus, sirolimus, everolimus, alfentanil, diergotamină, ergotamină, fentanil şi chinidină trebuie făcută cu precauţie (vezi pct. 4.4).
Corticosteroizi
Dexametazonă: În cazul administrării concomitente cu EMEND 125 mg/80 mg, doza orală uzuală de dexametazonă trebuie redusă cu aproximativ 50%. În studii clinice pentru chimioterapia inducătoare de greaţă şi vărsături (GVIC), alegerea dozei de dexametazonă a fost făcută în funcţie de interacţiunile cu alte substanţe active (vezi pct. 4.2). Administrarea EMEND în ziua 1 în doză de 125 mg în asociere cu 20 mg dexametazonă pe cale orală, iar în zilele 2-5 în doză de 80 mg/zi în asociere cu 8 mg dexametazonă pe cale orală, a crescut ASC pentru dexametazonă, un substrat al CYP3A4, de 2,2 ori în zilele 1 şi 5.
Metilprednisolonă: În cazul asocierii cu EMEND 125 mg/80 mg, doza uzuală de metilprednisolonă administrată pe cale intravenoasă trebuie redusă cu aproximativ 25%, iar doza uzuală de metilprednisolonă administrată pe cale orală trebuie redusă cu aproximativ 50%. EMEND, administrat în doză de 125 mg în ziua 1 şi de 80 mg/zi în zilele 2 şi 3, a crescut ASC pentru metilprednisolonă, un substrat pentru CYP3A4, de 1,3 ori în ziua 1 şi de 2,5 ori în ziua 3, atunci când aceasta a fost administrată concomitent, pe cale intravenoasă în doză de 125 mg în ziua 1 şi pe cale orală în doză de 40 mg în zilele 2 şi 3.
În timpul tratamentului continuu cu metilprednisolon, ASC pentru metilprednisolon poate scădea mai târziu, în intervalul de 2 săptămâni după iniţierea administrării EMEND, datorită efectului inductor al aprepitant asupra CYP3A4. Este de aşteptat ca acest efect să fie mai pronunţat în cazul administrării orale a metilprednisolonei.
Medicamente chimioterapice
În studii farmacocinetice, când a fost administrat în regim de 125 mg în ziua 1 şi 80 mg în zilele 2 şi 3, EMEND nu a influenţat farmacocinetica docetaxel administrat intravenos în ziua 1 sau a vinorelbin administrat intravenos în ziua 1 sau ziua 8. Deoarece efectul EMEND pe farmacocinetica administrării orale a substraturilor CYP3A4 este mai mare decât efectul EMEND pe farmacocinetica administrării intravenoase a substraturilor CYP3A4, interacţiunea cu medicamentele chimioterapice administrate oral, metabolizate în principal sau parţial de către CYP3A4 (de exemplu, etopozid, vinorelbin) nu poate fi exclusă. La pacienţii cărora li se administrează medicamente metabolizate în principal sau parţial de către CYP3A4 se recomandă precauţie şi monitorizare suplimentară (vezi pct. 4.4). După punerea pe piaţă, în timpul administrării concomitente de aprepitant cu ifosfamidă, au fost raportate evenimente de neurotoxicitate, o posibilă reacţie adversă a ifosfamidei.
Imunosupresoare
Pe durata regimului GVIC de 3 zile, este de aşteptat o creştere tranzitorie moderată urmată de o scădere uşoară a expunerii imunosupresoarelor metabolizate pe calea CYP3A4 (de exemplu, ciclosporină, tacrolimus, everolimus şi sirolimus). Datorită duratei scurte a regimului de 3 zile şi schimbărilor limitate, dependente de timp, privind expunerea, nu este recomandată reducerea dozei de imunosupresor pe durata celor 3 zile de administrare în asociere cu EMEND.
Midazolam
În cazul administrării concomitente a EMEND (125 mg/80 mg) cu midazolam sau alte benzodiazepine metabolizate pe calea CYP3A4 (alprazolam, triazolam), trebuie avute în vedere efectele posibilei creşteri a concentraţiilor plasmatice ale acestor medicamente.
EMEND a crescut ASC pentru midazolam, un substrat sensibil al CYP3A4, de 2,3 ori în ziua 1 şi de 3,3 ori în ziua 5, în cazul în care au fost administrate doze orale unice de 2 mg midazolam în zilele 1 şi 5 ale unei scheme terapeutice cu 125 mg EMEND în ziua 1 şi 80 mg/zi în zilele 2-5.
În alt studiu care a utilizat administrarea intravenoasă de midazolam, EMEND a fost administrat în doză de 125 mg în ziua 1 şi de 80 mg/zi în zilele 2 şi 3, iar midazolam a fost administrat intravenos în doză de 2 mg anterior regimului de 3 zile cu EMEND şi în zilele 4, 8 şi 15. EMEND a crescut ASC pentru midazolam cu 25% în ziua 4 şi a scăzut ASC pentru midazolam cu 19% în ziua 8 şi cu 4% în ziua 15. Aceste efecte nu au fost considerate importante din punct de vedere clinic.
Într-un al treilea studiu în care s-a administrat midazolam intravenos şi oral, EMEND a fost administrat în doză de 125 mg în ziua 1 şi de 80 mg/zi în zilele 2 şi 3, în asociere cu ondansetron 32 mg în ziua 1, dexametazonă 12 mg în ziua 1 şi 8 mg în zilele 2-4. Această asociere (EMEND, ondansetron şi dexametazonă) a scăzut ASC pentru midazolam administrat oral cu 16%în ziua 6, cu 9% în ziua 8, cu 7% în ziua 15 şi cu 17% în ziua 22. Aceste efecte nu au fost considerate importante din punct de vedere clinic.
S-a efectuat un studiu adițional privind administrarea intravenoasă de midazolam şi EMEND. Midazolam intravenos 2 mg a fost administrat la 1 oră după administrarea orală a unei doze unice de EMEND de 125 mg. ASC plasmatică a midazolamului a crescut de 1,5 ori. Acest efect nu a fost considerat important din punct de vedere clinic.
Inducţie
Fiind un inductor slab al CYP2C9, al CYP3A4 şi al glucuronoconjugării, aprepitantul poate reduce concentraţiile plasmatice ale substraturilor eliminate pe aceste căi timp de două săptămâni după iniţierea tratamentului. Acest efect poate deveni evident doar după terminarea tratamentului de 3 zile cu EMEND. Inducţia este tranzitorie în cazul substraturilor pentru CYP2C9 şi CYP3A4, iar efectul maxim este atins după 3-5 zile de la terminarea curei de 3 zile cu EMEND. Efectul se menţine câteva zile, ulterior se reduce progresiv şi devine clinic nesemnificativ după două săptămâni de la terminarea tratamentului cu EMEND. Inducţia uşoară a glucuronoconjugării este observată şi după administrarea orală a unei doze de 80 mg de aprepitant timp de 7 zile. Nu sunt disponibile date referitoare la efectele asupra CYP2C8 şi CYP2C19. Se recomandă precauţie la administrarea în această perioadă de timp a warfarinei, acenocumarolului, tolbutamidei, fenitoinei sau a altor substanţe active cunoscute a fi metabolizate pe calea CYP2C9.
Warfarină
La pacienţii aflați în tratament cronic cu warfarină, timpul de protrombină (INR) trebuie monitorizat cu atenţie în timpul tratamentului cu EMEND şi timp de 2 săptămâni după fiecare cură de 3 zile cu EMEND (vezi pct. 4.4). Când a fost administrată o doză unică de 125 mg EMEND în ziua 1 şi de 80 mg/zi în zilele 2 şi 3 la subiecţi sănătoşi, stabilizaţi pe o schemă de tratament cronic cu warfarină, nu s-a observat niciun efect al EMEND asupra ASC plasmatice a warfarinei R(+) sau S(-) determinată în ziua 3; cu toate acestea, s-a observat o reducere cu 34% a concentraţiei minime de warfarină S(-) (substrat al CYP2C9) însoţită de o reducere cu 14% a INR la 5 zile după terminarea tratamentului cu EMEND.
Tolbutamidă
În cazul administrării orale a unor doze unice de 500 mg tolbutamidă anterior regimului de 3 zile de EMEND şi în zilele 4, 8 şi 15, EMEND, administrat în doză de 125 mg în ziua 1 şi de 80 mg/zi în zilele 2 şi 3, a scăzut ASC pentru tolbutamidă (substrat al CYP2C9) cu 23% în ziua 4, cu 28% în ziua 8 şi cu 15% în ziua 15.
Contraceptivele hormonale
Eficacitatea contraceptivelor hormonale poate fi redusă în timpul administrării EMEND şi timp de 28 zile după aceea. Pe durata tratamentului cu EMEND şi timp de 2 luni după administrarea ultimei doze, trebuie utilizate metode contraceptive alternative non hormonale suplimentare.
Într-un studiu clinic au fost administrate doze unice dintr-un contraceptiv oral conținând etinilestradiol şi noretindronă în zilele 1-21 împreună cu EMEND 125 mg în ziua 8 şi 80 mg/zi în zilele 9 şi 10, ondansetron 32 mg intravenos în ziua 8 şi dexametazonă administrată oral în doze de 12 mg în ziua 8 şi 8 mg/zi în zilele 9, 10 şi 11. În zilele 9-21 ale acestui studiu, s-au înregistrat reduceri de până la 64% ale concentraţiilor bazale de etinilestradiol şi reduceri de până la 60% ale concentraţiilor bazale de noretindronă.
Antagonişti 5-HT3
În studiile clinice privind interacţiunea, aprepitant nu a prezentat efecte importante din punct de vedere clinic asupra farmacocineticii pentru ondansetron, granisetron sau hidrodolasetron (metabolitul activ al dolasetronului).
Efectul altor medicamente asupra farmacocineticii aprepitantului
Administrarea concomitentă de EMEND cu substanţe active care inhibă activitatea CYP3A4 (de exemplu, ketoconazol, itraconazol, voriconazol, posaconazol, claritromicină, telitromicină, nefazodonă şi inhibitori de protează) trebuie efectuată cu prudenţă, deoarece este de aşteptat ca asocierea să determine creşterea de câteva ori a concentraţiilor plasmatice de aprepitant (vezi pct. 4.4).
Administrarea concomitentă a EMEND cu substanţe active puternic inductoare ale activităţii CYP3A4 (de exemplu, rifampicină, fenitoină, carbamazepină, fenobarbital) trebuie evitată deoarece asocierea determină reducerea concentraţiilor plasmatice de aprepitant, cu posibila reducere a eficacităţii EMEND. Nu se recomandă administrarea concomitentă de EMEND cu preparate din plante medicinale ce conţin sunătoare (Hypericum perforatum).
Ketoconazol
La administrarea unei doze unice de 125 mg aprepitant în ziua 5 a unui regim terapeutic de 10 zile cu ketoconazol în doză de 400 mg/zi, inhibitor puternic al CYP3A4, ASC pentru aprepitant a crescut de aproximativ 5 ori şi timpul mediu terminal de înjumătăţire al aprepitantului a crescut de aproximativ 3 ori.
Rifampicină
La administrarea unei doze unice de 375 mg aprepitant în ziua 9 a unui regim terapeutic de 14 zile cu rifampicină în doză de 600 mg/zi, inductor puternic al CYP3A4, ASC pentru aprepitant a scăzut cu 91% şi timpul mediu terminal de înjumătăţire a scăzut cu 68%.
Copii și adolescenți
Au fost efectuate studii privind interacțiunile numai la adulți.
4.6 Fertilitatea, sarcina şi alăptarea
Contracepţia la bărbaţi şi femei
Eficacitatea contraceptivelor hormonale poate fi redusă pe perioada administrării şi timp de 28 zile după administrarea EMEND. Trebuie folosite metode contraceptive alternative non hormonale de rezervă în timpul tratamentului cu EMEND şi timp de 2 luni după ultima doză de EMEND (vezi pct. 4.4 şi 4.5).
Sarcina
Nu sunt disponibile date clinice privind expunerea sarcinilor la aprepitant. Nu s-a stabilit complet potenţialul toxicității reproductive a aprepitantului, deoarece în studiile la animale nu au putut fi atinse nivelurile de expunere superioare celor ale expunerii umane la doza de 125 mg/80 mg. Aceste studii nu au evidenţiat efecte nocive directe sau indirecte asupra sarcinii, dezvoltării embrionare/fetale, naşterii sau dezvoltării postnatale (vezi pct. 5.3). Nu se cunosc efectele potenţiale asupra reproducerii modificărilor în reglarea neurokininei. EMEND nu trebuie administrat în timpul sarcinii decât dacă este absolut necesar.
Alăptarea
Aprepitant este excretat în laptele femelelor de şobolani. Nu se cunoaşte dacă aprepitant este excretat în laptele uman; de aceea, alăptarea nu este recomandată în timpul tratamentului cu EMEND.
Fertilitatea
Nu s-a stabilit în mod cert potenţialul aprepitantului de a afecta fertilitatea, deoarece în studiile la animale nu au putut fi atinse nivelurile de expunere superioare celor ale expunerii umane la doza terapeutică. Aceste studii de fertilitate nu au evidenţiat efecte nocive directe sau indirecte asupra capacităţii de reproducere, fertilităţii, dezvoltării embrionare/fetale sau numărului şi motilităţii spermatozoizilor (vezi pct. 5.3).
4.7 Efecte asupra capacităţii de a conduce vehicule şi de a folosi utilaje
EMEND poate avea o influenţă minoră asupra capacităţii de a merge cu bicicleta şi de a folosi utilaje. După administrarea EMEND pot să apară ameţeală şi fatigabilitate (vezi pct. 4.8).
4.8 Reacţii adverse
Rezumatul profilului de siguranţă
Profilul de siguranţă al aprepitantului a fost evaluat la aproximativ 6500 adulţi în mai mult de 50 studii şi 184 copii şi adolescenţi din 2 studii clinice pediatrice pivot.
Cele mai frecvente reacţii adverse raportate cu o incidenţă mai mare la adulții trataţi cu o schemă terapeutică pe bază de aprepitant, comparativ cu cei cărora li s-a administrat terapie standard, în cursul Chimioterapiei cu Potenţial Emetogen Crescut (CPEC), au fost: sughiţ (4,6% versus 2,9%), creşterea alanin aminotransferazei (ALAT) (2,8% versus 1,1%), dispepsie (2,6% versus 2,0%), constipaţie (2,4% versus 2,0 %), cefalee (2,0% versus 1,8%) şi scăderea apetitului alimentar (2,0% versus 0,5%). Cea mai frecventă reacţie adversă raportată cu o incidenţă mai mare la pacienţii trataţi cu o schemă terapeutică pe bază de aprepitant, comparativ cu adulții cărora li s-a administrat tratament standard, în cursul Chimioterapiei cu Potenţial Emetogen Moderat (CPEM) a fost fatigabilitatea (1,4% versus 0,9%).
În timpul administrării chimioterapiei anticanceroase cu potențial emetogen, cele mai frecvente reacții adverse raportate cu o incidență mai mare la copii și adolescenți tratați urmând o schemă terapeutică pe bază de aprepitant comparativ cu schema terapeutică de control au fost sughiț (3,3 % comparativ cu 0,0 %) și hiperemie (1,1 % comparativ cu 0,0 %).
Lista reacţiilor adverse în format tabelar
Următoarele reacţii adverse au fost observate într-o analiză cumulată a studiilor CPEC şi CPEM cu o incidenţă mai mare la aprepitant decât la terapia standard sau în utilizarea după punerea pe piaţă. Categoriile de frecvență din tabel se bazează pe studiile la adulți; frecvențele observate în studiile la copii și adolescenți au fost similare sau mai scăzute, cu excepția cazurilor prezentate în tabel. Unele RA mai puțin frecvente la populația adultă nu au fost observate în studiile la copii și adolescenți.
Frecvenţele sunt definite astfel: foarte frecvente (≥ 1/10); frecvente (≥ 1/100 şi < 1/10); mai puţin frecvente (≥ 1/1000 şi < 1/100); rare (≥ 1/10000 şi < 1/1000) şi foarte rare (< 1/10000), cu frecvenţă necunoscută (care nu poate fi estimată din datele disponibile).
| Clasificare pe aparate, sisteme şi organe | Reacţia adversă | Frecvenţa |
|---|---|---|
| Infecţii şi infestări | candidoză, infecţie stafilococică | rare |
| Tulburări hematologice şi limfatice | neutropenie febrilă, anemie | mai puţin frecvente |
| Tulburări ale sistemului imunitar | reacţii de hipersensibilitate incluzând reacţii anafilactice | necunoscută |
| Tulburări metabolice şi de nutriţie | scăderea apetitului alimentar | frecvente |
| polidipsie | rare | |
| Tulburări psihice | anxietate | mai puţin frecvente |
| dezorientare, dispoziţie euforică | rare | |
| Tulburări ale sistemului nervos | cefalee | frecvente |
| ameţeală, somnolenţă | mai puţin frecvente | |
| tulburare cognitivă, letargie, disgeuzie | rare | |
| Tulburări oculare | conjunctivită | rare |
| Tulburări acustice şi vestibulare | tinitus | rare |
| Tulburări cardiace | palpitaţii | mai puţin frecvente |
| bradicardie, tulburări cardiovasculare | rare | |
| Tulburări vasculare | bufeuri/hiperemie | mai puţin frecvente |
| Tulburări respiratorii, toracice şi mediastinale | sughiţ | frecvente |
| durere orofaringiană, strănut, tuse, creșterea secrețiilor nazale, iritaţie faringiană | rare | |
| Tulburări gastro-intestinale | constipaţie, dispepsie | frecvente |
| eructaţie, greaţă†, vărsături†, boală de reflux gastro-esofagian, durere abdominală, xerostomie, flatulenţă | mai puţin frecvente | |
| ulcer duodenal perforat, stomatită, distensie abdominală, materii fecale tari, colită neutropenică | rare | |
| Tulburări cutanate şi ale ţesutului subcutanat | erupţii cutanate tranzitorii, acnee | mai puţin frecvente |
| reacţie de fotosensibilitate, hiperhidroză, seboree, leziuni cutanate, erupţii cutanate tranzitorii pruriginoase, sindrom Stevens-Johnson/necroliză epidermică toxică | rare | |
| prurit, urticarie | necunoscută | |
| Tulburări musculo-scheletice şi ale ţesutului conjunctiv | slăbiciune musculară, spasme musculare | rare |
| Tulburări renale şi ale căilor urinare | disurie | mai puţin frecvente |
| polakiurie | rare | |
| Tulburări generale şi la nivelul locului de administrare | fatigabilitate | frecvente |
| astenie, stare de rău general | mai puţin frecvente | |
| edeme, disconfort toracic, tulburări de mers | rare | |
| Investigaţii diagnostice | creşterea ALAT | frecvente |
| creşterea ASAT, creşterea fosfatazei alcaline | mai puţin frecvente | |
| hematurie, hiponatremie, scădere ponderală, scăderea numărului de neutrofile, glucozurie, eliminare crescută de urină | rare |
†Greaţa şi vărsăturile au reprezentat parametrii de eficacitate în primele 5 zile după tratamentul chimioterapic şi au fost raportate ca reacţii adverse numai după aceea.
Descrierea reacţiilor adverse selectate
Profilurile reacţiilor adverse la adulți pentru extensia Multiple-Cycle (cicluri multiple) din studiile CPEC şi CPEM de până la 6 cicluri suplimentare de chimioterapie au fost, în general, similare cu cele 1.
Într-un studiu clinic suplimentar controlat cu comparator activ la 1169 pacienţi cărora li s-a administrat aprepitant şi CPEC, profilul reacţiilor adverse a fost în general similar celui observat în alte studii CPEC cu aprepitant.
Studii non-GVIC
Reacţii adverse suplimentare au fost observate la pacienţii adulți trataţi cu o doză unică de 40 mg de aprepitant pentru starea de starea de greaţă şi vărsăturile postoperatorii (GVPO) cu o incidenţă mai mare decât cu ondansetron: dureri în etajul abdominal superior, sunete intestinale anormale, constipaţie*, disartrie, dispnee, hipoestezie, insomnie, mioză, greaţă, perturbări senzoriale, disconfort stomacal, subileus*, acuitate vizuală redusă, wheezing.
*Raportate la pacienţii care utilizează o doză mai mare de aprepitant.
Raportarea reacţiilor adverse suspectate
Raportarea reacţiilor adverse suspectate după autorizarea medicamentului este importantă. Acest lucru permite monitorizarea continuă a raportului beneficiu/risc al medicamentului. Profesioniştii din domeniul sănătăţii sunt rugaţi să raporteze orice reacţie adversă suspectată prin intermediul sistemului naţional de raportare, astfel cum este menţionat în Anexa V.
4.9 Supradozaj
În cazul unui supradozaj trebuie întreruptă administrarea EMEND şi trebuie efectuat tratament general de susţinere a funcţiilor vitale şi monitorizare. Datorită acţiunii antiemetice a aprepitantului, este posibil ca emeza indusă de un medicament să nu fie eficientă în aceste cazuri.
Aprepitant nu poate fi eliminat prin hemodializă.
5. PROPRIETĂŢI FARMACOLOGICE
5.1 Proprietăţi farmacodinamice
Grupa farmacoterapeutică: Antiemetice şi medicamente pentru combaterea greţei, codul ATC: A04AD12
Aprepitant este un antagonist selectiv, cu afinitate înaltă, al receptorilor neurokininici 1 (NK1) pentru substanţa P umană.
Schema terapeutică de 3 zile pe bază de aprepitant la adulţi
În 2 studii randomizate, de tip dublu-orb, care au cuprins un număr total de 1094 pacienţi adulți trataţi cu chimioterapie care a inclus cisplatină în doză ≥ 70 mg/m2, administrarea aprepitantului în asociere cu o schemă terapeutică de ondansetron/dexametazonă (vezi pct. 4.2) a fost comparată cu o schemă terapeutică standard (placebo plus ondansetron 32 mg administrat intravenos în ziua 1 plus dexametazonă administrată pe cale orală 20 mg în ziua 1 şi 8 mg de 2 ori pe zi în zilele 2-4). Deşi în studiile clinice a fost folosită doza de 32 mg ondansetron administrat intravenos, aceasta nu mai este doza recomandată. A se vedea informaţiile despre medicament pentru antagonistul receptorilor 5-HT3 selectat pentru informaţii corespunzătoare privind dozarea.
Evaluarea eficacităţii s-a bazat pe următorul obiectiv compus: răspuns complet (definit ca absenţa episoadelor emetice şi absenţa administrării terapiei de salvare), în principal în timpul ciclului 1. Rezultatele au fost evaluate pentru fiecare studiu în mod individual şi pentru cele 2 studii combinate.
În Tabelul 1 este prezentat un rezumat al principalelor rezultate ale analizei combinate ale celor două studii.
Tabelul 1
Procentul răspunsurilor la pacienţii adulți la care s-a administrat chimioterapie cu potenţial emetogen crescut, pe grupe de tratament şi fază — Ciclul 1
Schemă Tratament Diferenţe* terapeutică standard
OBIECTIVE COMPUSE pe bază de (N= 524)† aprepitant % % (IÎ 95%) (N= 521)†
%
| Răspuns complet (fără emeză şi fără terapie de salvare) | ||||
|---|---|---|---|---|
| Global (0-120 ore) | 67,7 | 47,8 | 19,9 | (14,0, 25,8) |
| 0-24 ore | 86,0 | 73,2 | 12,7 | (7,9, 17,6) |
| 25-120 ore | 71,5 | 51,2 | 20,3 | (14,5, 26,1) |
| OBIECTIVE INDIVIDUALE | ||||
| Fără emeză (fără episoade de emeză, indiferent de utilizarea sau nu a terapiei de salvare) | ||||
| Global (0-120 ore) | 71,9 | 49,7 | 22,2 | (16,4, 28,0) |
| 0-24 ore | 86,8 | 74,0 | 12,7 | (8,0, 17,5) |
| 25-120 ore | 76,2 | 53,5 | 22,6 | (17,0, 28,2) |
| Fără greţuri semnificative (VAS maxim < 25 mm pe o scală între 0-100 mm) | ||||
| Global (0-120 ore) | 72,1 | 64,9 | 7,2 | (1,6, 12,8) |
| 25-120 ore | 74,0 | 66,9 | 7,1 | (1,5, 12,6) |
* Intervalele de încredere au fost calculate fără ajustări în funcţie de sex şi chimioterapia concomitentă, care au fost incluse în analiza iniţială a riscului relativ şi a modelelor logistice.
† Un pacient din schema terapeutică pe bază de aprepitant a prezentat date doar în faza acută şi a fost exclus din analizele globale din faza finală; un pacient din schema terapeutică standard a prezentat date doar în faza finală şi a fost exclus din analizele globale din faza acută.
Intervalul estimat până la prima emeză în analiza combinată este ilustrat prin curba Kaplan-Meier din Figura 1.
Figura 1
Procentul de pacienţi adulți trataţi cu chimioterapie cu potenţial emetogen crescut care nu a prezentat emeză pe intervalul de timp – Ciclul 1
100%
Schemă terapeutică cu aprepitant (N=520) Terapie standard (N=523)
90%
Procent de pacienţi
80%
70%
60%
50%
40%
0
0 36 72 108
12 24 48 60 84 96 120
Timp (ore)
Diferenţe semnificative statistic în ceea ce priveşte eficacitatea au fost, de asemenea, observate în fiecare dintre cele 2 studii analizate individual.
În aceleaşi 2 studii clinice, 851 pacienţi adulți au continuat tratamentul în extensia Multiple Cycle (cicluri multiple) până la 5 cicluri suplimentare de chimioterapie. Aparent, eficacitatea schemei terapeutice pe bază de aprepitant s-a menţinut pe durata tuturor ciclurilor.
Într-un studiu randomizat, dublu-orb, care a cuprins un număr total de 866 pacienţi adulţi (864 femei, 2 bărbaţi) trataţi cu chimioterapie care a inclus ciclofosfamidă 750-1500 mg/m2; sau ciclofosfamidă 500-1500 mg/m2 şi doxorubicină (≤ 60 mg/m2) sau epirubicină (≤ 100 mg/m2), administrarea aprepitantului în asociere cu o schemă terapeutică cu ondansetron/dexametazonă (vezi pct. 4.2) a fost comparată cu tratamentul standard (placebo plus ondansetron 8 mg administrat pe cale orală de 2 ori pe zi în ziua 1 şi la fiecare 12 ore în zilele 2 şi 3) plus dexametazonă 20 mg administrată pe cale orală în ziua 1.
Evaluarea eficacităţii s-a bazat pe un obiectiv compus: răspuns complet (definit ca absenţa episoadelor emetice şi absenţa administrării terapiei de salvare), în principal în timpul ciclului 1.
Un rezumat al principalelor rezultate ale studiului este prezentat în Tabelul 2.
Tabelul 2
Procentul răspunsurilor la pacienţii adulți pe grupe de tratament şi fază — Ciclul 1 Chimioterapie cu potenţial emetogen moderat
Schemă Tratament Diferenţe* terapeutică standard
OBIECTIVE COMPUSE pe bază de (N= 424) aprepitant % % (IÎ 95%) (N= 433)†
%
| Răspuns complet (fără emeză şi fără terapie de salvare) | ||||
|---|---|---|---|---|
| Global (0-120 ore) 0-24 ore 25-120 ore OBIECTIVE INDIVIDUALE | 50,8 75,7 55,4 | 42,5 69,0 49,1 | 8,3 6,7 6,3 | (1,6, 15,0) (0,7, 12,7) (-0,4, 13,0) |
| Fără emeză (fără episoade de emeză, indiferent de utilizarea sau nu a terapiei de salvare) | ||||
| Global (0-120 ore) 0-24 ore 25-120 ore | 75,7 87,5 80,8 | 58,7 77,3 69,1 | 17,0 10,2 11,7 | (10,8, 23,2) (5,1, 15,3) (5,9, 17,5) |
| Fără greţuri semnificative (VAS maxim < 25 mm pe o scală între 0-100 mm) | ||||
| Global (0-120 ore) | 60,9 | 55,7 | 5,3 | (-1,3, 11,9) |
| 0-24 ore | 79,5 | 78,3 | 1,3 | (-4,2, 6,8) |
| 25-120 ore | 65,3 | 61,5 | 3,9 | (-2,6, 10,3) |
* Intervalele de încredere au fost calculate fără ajustări în funcţie de grupa de vârstă (<55 ani, ≥55 ani) şi de grupul investigator, care au fost incluse în analiza iniţială a riscului relativ şi a modelelor logistice. † Un pacient din schema terapeutică pe bază de aprepitant a prezentat date doar în faza acută şi a fost exclus din analizele globale din faza finală.
În acelaşi studiu clinic, 744 pacienţi adulți au continuat tratamentul în extensia Multiple Cycle (cicluri multiple) până la 3 cicluri suplimentare de chimioterapie. Aparent, eficacitatea schemei terapeutice pe bază de aprepitant s-a menţinut pe durata tuturor ciclurilor.
Într-un al doilea studiu clinic multicentric, randomizat, dublu-orb, cu grupuri paralele, schema terapeutică pe bază de aprepitant a fost comparată cu schema terapeutică standard la 848 pacienţi (652 femei, 196 bărbaţi) trataţi cu o schemă chimioterapeutică care a inclus oricare doză intravenoasă de oxaliplatin, carboplatin, epirubicină, idarubicină, ifosfamidă, irinotecan, daunorubicină, doxorubicină; ciclofosfamidă intravenoasă (< 1500 mg/m2); sau citarabină intravenoasă (> 1 g/m2). Pacienţilor trataţi cu schema terapeutică pe bază de aprepitant li s-a administrat chimioterapie pentru o varietate de tipuri de tumori incluzând 52% cu cancer de sân, 21% cu cancere gastro-intestinale inclusiv cancer colorectal, 13% cu cancer pulmonar şi 6% cu cancere ginecologice. Schema terapeutică pe bază de aprepitant în asociere cu o schemă terapeutică cu ondansetron/dexametazonă (vezi pct. 4.2) a fost comparată cu terapia standard (placebo în asociere cu ondansetron 8 mg administrat pe cale orală (de 2 ori pe zi în ziua 1 şi la intervale de 12 ore în zilele 2 şi 3) plus dexametazonă 20 mg administrată pe cale orală în ziua 1).
Evaluarea eficacităţii s-a bazat pe următoarele criterii principale şi secundare finale de evaluare: absenţa vărsăturilor în perioada globală (0 până la 120 ore post-chimioterapie), evaluarea siguranţei şi tolerabilităţii schemei terapeutice pe bază de aprepitant pentru greaţa şi vărsăturile induse de chimioterapie (GVIC), şi răspuns complet (definit ca absenţa vărsăturilor şi absenţa administrării terapiei de salvare) în perioada globală (0 până la 120 ore post-chimioterapie). În plus, absenţa greţei semnificative în perioada globală (0 până la 120 ore post-chimioterapie) a fost evaluată ca un criteriu explorator final de evaluare şi în fazele acute şi întârziate ca analiză post-hoc.
Un rezumat al principalelor rezultate ale studiului este prezentat în Tabelul 3.
Tabelul 3
Procentul răspunsurilor la pacienţii adulți pe grupe de tratament şi fază pentru Studiul 2 — Ciclul 1 Chimioterapie cu potenţial emetogen moderat
| Schemă terapeutică pe bază de aprepitant (N= 425) % | Tratament standard (N= 406) % | Diferenţe* % (IÎ 95%) | ||
| Răspuns complet (fără emeză şi fără terapie de salvare) | ||||
|---|---|---|---|---|
Global (0-120 ore) 0-24 ore 25-120 ore | 68,7 89,2 70,8 | 56,3 80,3 60,9 | 12,4 8,9 9,9 | (5,9, 18,9) (4,0, 13,8) (3,5, 16,3) |
| Fără emeză (fără episoade de emeză, indiferent de utilizarea sau nu a terapiei de salvare) | ||||
Global (0-120 ore) 0-24 ore 25-120 ore | 76,2 92,0 77,9 | 62,1 83,7 66,8 | 14,1 8,3 11,1 | (7,9, 20,3) (3,9, 12,7) (5,1, 17,1) |
| Fără greţuri semnificative (VAS maxim < 25 mm pe o scală între 0-100 mm) | ||||
Global (0-120 ore) 0-24 ore 25-120 ore | 73,6 90,9 74,9 | 66,4 86,3 69,5 | 7,2 4,6 5,4 | (1,0, 13,4) (0,2, 9,0) (-0,7, 11,5) |
*Intervalele de încredere au fost calculate fără ajustări în funcţie de sex şi regiune, care au fost incluse în analiza iniţială utilizând modelele logistice.
Beneficiul schemei terapeutice asociate cu aprepitant la întreaga populaţie participantă la studiu a fost determinat în principal de rezultatele observate la pacienţii cu un control slab obţinut cu schema terapeutică standard, cum ar fi la femei, deşi rezultatele au fost numeric mai bune indiferent de vârstă, tip de tumoră sau sex. A fost obţinut răspuns complet la schema terapeutică pe bază de aprepitant şi respectiv la tratamentul standard la 209/304 (65%) şi 161/320 (50%) la femei şi 83/101 (82%) şi 68/87 (78%) la bărbaţi.
Copii şi adolescenţi
Într-un studiu clinic randomizat, dublu-orb, controlat cu comparator activ, care a inclus 302 copii şi adolescenţi (cu vârsta cuprinsă între 6 luni şi 17 ani) cărora li s-au administrat chimioterapie cu potențial emetogen moderat sau mare, schema terapeutică pe bază de aprepitant a fost comparată cu o schemă terapeutică de control pentru prevenirea GVIC. Eficacitatea schemei terapeutice cu aprepitant a fost evaluată într-un singur ciclu (Ciclul 1). Pacienţii au avut posibilitatea de a urma în mod deschis tratamentul cu aprepitant în cicluri ulterioare (Cicluri opționale 2-6); cu toate acestea, eficacitatea nu a fost evaluată în aceste cicluri opţionale. Schema terapeutică pe bază de aprepitant pentru adolescenţii cu vârsta cuprinsă între 12 şi 17 ani (n=47) a constat în EMEND capsule 125 mg pe cale orală în ziua 1 şi 80 mg/zi în zilele 2 şi 3, în asociere cu ondansetron în ziua 1. Schema terapeutică pe bază de aprepitant pentru copiii cu vârsta cuprinsă între 6 luni şi 12 ani (n=105) a constat în EMEND pulbere pentru suspensie orală 3,0 mg/kg (până la 125 mg) pe cale orală în ziua 1 şi 2,0 mg/kg (până la 80 mg) pe cale orală în zilele 2 şi 3, în asociere cu ondansetron în ziua 1. Schema terapeutică de control la adolescenţii cu vârsta cuprinsă între 12 şi 17 ani (n=48) și copiii cu vârsta cuprinsă între 6 luni şi 12 ani (n=102) a constat în placebo în loc de aprepitant în zilele 1, 2 şi 3 în asociere cu ondansetron în ziua 1. EMEND sau placebo şi ondansetron au fost administrate cu 1 oră şi respectiv cu 30 minute înainte de iniţierea chimioterapiei. Administrarea intravenoasă a dexametazonei a fost permisă în cadrul schemei terapeutice antiemetice pentru copiii și adolescenții din ambele grupe de vârstă, în funcție de decizia medicului. În cazul copiilor și adolescenților care au primit aprepitant, a fost necesară reducerea dozei (50%) de dexametazonă. Nu a fost necesară reducerea dozei în cazul copiilor și adolescenților care au primit schema terapeutică de control. Dintre copii și adolescenți, la 29% din cei care au primit schema terapeutică pe bază de aprepitant şi la 28% dintre cei care au primit schema terapeutică de control s-a administrat dexametazona ca parte a schemei terapeutice din Ciclul 1.
Activitatea antiemetică a EMEND a fost evaluată pe o perioadă de 5 zile (120 ore) după iniţierea chimioterapiei în ziua 1. Obiectivul clinic primar a fost răspunsul complet în faza tardivă (25 până la 120 ore după iniţierea chimioterapiei) în Ciclul 1. Un rezumat al principalelor rezultate ale studiului este prezentat în Tabelul 4.
Tabelul 4
Numărul (%) de copii și adolescenți cu răspuns complet şi fără vărsături pe grupe de tratament şi fază
- Ciclul 1 (populaţie în intenţie de tratament)
| Schema terapeutică pe bază de aprepitant n/m (%) | Schema terapeutică de control n/m (%) | |
| OBIECTIV CLINIC PRIMAR | ||
|---|---|---|
| Răspuns complet*– Faza tardivă | 77/152 (50,7)† | 39/150 (26,0) |
| ALTE OBIECTIVE CLINICE PRESPECIFICATE | ||
| Răspuns complet*– Faza acută | 101/152 (66,4)‡ | 78/150 (52,0) |
| Răspuns complet*– Faza globală | 61/152 (40,1)† | 30/150 (20,0) |
| Fără vărsături§– Faza globală | 71/152 (46,7)† | 32/150 (21,3) |
*Răspuns complet = Fără vărsături sau eructație sau reflex de vărsăturăşi fără utilizarea medicaţiei de urgență. †p < 0,01 atunci când se comparăcu schema terapeutică de control. ‡p < 0,05 atunci când se comparaă cu schema terapeutică de control. §Fără vărsături = Fără vărsături sau eructație sau reflex de vărsătură. n/m = Număr de pacienţi cu răspunsul dorit/număr de pacienţi incluşi în interval. Faza acută: 0până la 24 ore dupăiniţierea chimioterapiei. Faza tardivă: 25până la 120 ore dupăiniţierea chimioterapiei. Faza globală: 0până la 120 ore dupăiniţierea chimioterapiei. | ||
Timpul estimat până la primele vărsături după iniţierea tratamentului chimioterapic a fost mai mare în cazul schemei terapeutice pe bază de aprepitant (timpul median estimat până la primele vărsături a fost de 94,5 ore) comparativ cu grupul care a primit schema terapeutică de control (timpul median estimat până la primele vărsături a fost de 26,0 ore), așa cum arată curbele Kaplan-Meier din Figura 2.
Figura 2
Timpul până la primul episod de vărsături de la începutul administrării chimioterapiei - copii și adolescenți în faza globală-Ciclul 1 (populaţie în intenţie de tratament)
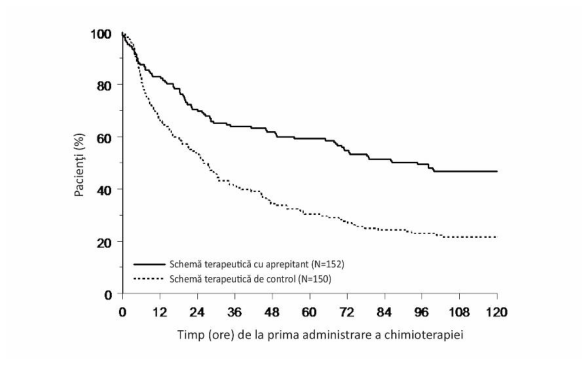
O analiză a eficacității la nivelul subpopulaţiilor din Ciclul 1 a demonstrat că, indiferent de categoria de vârstă, sex, utilizarea dexametazonei pentru profilaxia antiemetică şi de potenţialul emetogen al chimioterapiei, schema terapeutică pe bază de aprepitant a oferit un control mai bun faţă de cel obţinut cu schema terapeutică de control în ceea ce priveşte obiectivele clinice de răspuns complet.
5.2 Proprietăţi farmacocinetice
Aprepitant prezintă o farmacocinetică nelineară. Atât clearance-ul, cât şi biodisponibilitatea absolută scad odată cu creşterea dozei.
Absorbţie
Biodisponibilitatea absolută medie a aprepitantului după administrare orală este de 67% pentru capsula de 80 mg şi de 59% pentru capsula de 125 mg. Media concentraţiei plasmatice maxime (Cmax) a aprepitantului s-a înregistrat la aproximativ 4 ore (tmax). Administrarea orală a unei capsule cu un mic dejun standard de aproximativ 800 Kcal a determinat o creştere de până la 40% a ASC pentru aprepitant. Această creştere nu este considerată semnificativă din punct de vedere clinic.
Farmacocinetica aprepitantului este nelineară în limitele dozelor clinice. La adulţi tineri sănătoşi, creşterea ASC0-∞ a fost cu 26% mai mare decât raportul dozelor unice de 80 mg şi 125 mg administrate postprandial.
După administrarea pe cale orală a unei doze unice de 125 mg EMEND în ziua 1 şi 80 mg o dată pe zi în zilele 2 şi 3, ASC0-24ore (medie ± DS) a fost de 19,6 ± 2,5 µg•oră/ml şi 21,2 ± 6.3 µg •oră/ml în zilele 1 şi respectiv 3. Cmax a fost 1,6 ± 0,36 µg/ml şi 1,4 ± 0,22 µg/ml în zilele şi respectiv 3.
Distribuţie
Aprepitantul se leagă în proporţie mare de proteine, media fiind de 97%. La om, la starea de echilibru, volumul geometric aparent mediu de distribuţie (Vdss) este de aproximativ 66 l.
Metabolizare
Aprepitantul este supus unei metabolizări extensive. După administrarea intravenoasă a unei doze unice de 100 mg fosaprepitant marcat cu [14C], un promedicament pentru aprepitant, la adulţi tineri sănătoşi, aprepitantul este responsabil pentru aproximativ 19% din radioactivitatea plasmatică determinată după 72 ore, ceea ce indică prezenţa substanţială a metaboliţilor în plasmă. În plasma umană au fost identificaţi 12 metaboliţi ai aprepitantului. Metabolizarea aprepitantului se produce în principal prin oxidarea inelului morfolinic şi a catenelor sale, metaboliţii rezultaţi având o activitate slabă. Studiile in vitro care au utilizat microzomi hepatici umani indică faptul că aprepitantul este metabolizat în principal de către CYP3A4, cu o contribuţie potenţială minoră a CYP1A2 şi CYP2C19.
Eliminare
Aprepitantul nu se elimină nemetabolizat prin urină. Metaboliţii sunt excretaţi în urină, iar pe calea excreţiei biliare, în fecale. După administrarea intravenoasă la subiecţi sănătoşi a unei doze unice de 100 mg fosaprepitant marcat cu [14C], un promedicament pentru aprepitant, 57% din radioactivitate a fost detectată în urină, iar 45% în materiile fecale.
Clearance-ul plasmatic al aprepitantului este dependent de doză, scăzând cu creşterea acesteia şi variind între aproximativ 60 şi 72 ml/minut în intervalul dozelor terapeutice. Timpul terminal de înjumătăţire variază aproximativ de la 9 la 13 ore.
Farmacocinetica la grupe speciale de pacienţi
Sex: După administrarea orală a unei doze unice de 125 mg aprepitant, Cmax pentru aprepitant este cu 16% mai mare la femei, comparativ cu bărbaţii. Timpul de înjumătăţire al aprepitantului este cu 25% mai mic la femei comparativ cu bărbaţii, iar tmax se înregistrează după aproximativ acelaşi interval de timp. Aceste diferenţe nu au fost considerate semnificative din punct de vedere clinic. Nu este necesară ajustarea dozelor de EMEND în funcţie de sex.
Insuficienţă hepatică: Insuficienţa hepatică uşoară (Child-Pugh clasă A) nu influenţează farmacocinetica aprepitantului în mod semnificativ din punct de vedere clinic. Nu este necesară ajustarea dozei la pacienţii cu insuficienţă hepatică uşoară. Din datele existente nu pot fi trase concluzii referitoare la influenţa insuficienţei hepatice moderate (Child-Pugh clasă B) asupra farmacocineticii aprepitantului. Nu există date clinice sau farmacocinetice la pacienţii cu insuficienţă hepatică severă (Child-Pugh clasă C).
Insuficienţă renală: La pacienţi cu insuficienţă renală severă (Cl creatinină < 30 ml/minut) şi la cei cu afecţiuni renale în stadiu terminal (ARST) care necesită hemodializă a fost administrată o doză unică de 240 mg aprepitant.
La pacienţii cu insuficienţă renală severă, ASC0-∞ pentru aprepitantul total (liber şi nelegat de proteine) a scăzut cu 21%, iar Cmax a scăzut cu 32% comparativ cu subiecţii sănătoşi. La pacienţii cu ARST care fac hemodializă, ASC0-∞ pentru aprepitantul total a scăzut cu 42%, iar Cmax a scăzut cu 32%. Datorită scăderii uşoare a legării aprepitantului de proteine la pacienţii cu afecţiuni renale, ASC pentru aprepitantul liber, farmacologic activ, nu a fost influenţată semnificativ la pacienţii cu insuficienţă renală, comparativ cu subiecţii sănătoşi. Hemodializa efectuată la 4 sau 48 ore după administrarea dozei nu a avut efecte semnificative asupra farmacocineticii aprepitantului; mai puţin de 0,2% din doză a fost recuperată în dializat.
Nu este necesară ajustarea dozei de EMEND la pacienţi cu insuficienţă renală sau la pacienţi cu ARST care fac hemodializă.
Copii şi adolescenţi: În cadrul schemei terapeutice de 3 zile, dozarea capsulelor de aprepitant (125/80/80 mg) la pacienţi adolescenţi (cu vârsta cuprinsă între 12 şi 17 ani) a determinat o ASC0-24ore peste 17 µg•oră/ml în ziua 1, atingând concentraţii (Cmin) la sfârşitul zilelor 2 şi 3 peste 0,4 µg/ml la majoritatea pacienţilor. Valoarea mediană a concentraţiei plasmatice maxime (Cmax) a fost aproximativ 1,3 µg/ml în ziua 1, înregistrându-se la aproximativ 4 ore. În cadrul schemei terapeutice de 3 zile, dozarea pulberii pentru suspensie orală de aprepitant (3/2/2 mg/kg) la pacienţi cu vârsta cuprinsă între 6 luni şi 12 ani a determinat o ASC0-24ore peste 17 µg•oră/ml în ziua 1, cu concentraţii (Cmin) la sfârşitul zilelor 2 şi 3 şi peste 0,1 µg/ml la majoritatea pacienţilor. Valoarea mediană a concentraţiei plasmatice maxime (Cmax) a fost aproximativ 1,2 µg/ml în ziua 1, înregistrându-se între 5 și 7 ore.
O analiză farmacocinetiăi populaţională a aprepitantului la copii şi adolescenţi (cu vârsta cuprinsă între 6 luni şi 17 ani) sugerează faptul că sexul şi rasa nu au un efect semnificativ din punct de vedere clinic asupra farmacocineticii aprepitantului.
Relaţia între concentraţie şi efect
Utilizând un trasor cu specificitate crescută pentru receptorii NK1, studiile tomografice cu emisie de pozitroni (PET) efectuate la bărbaţi tineri sănătoşi au demonstrat faptul că aprepitantul pătrunde în creier şi se leagă de receptorii NK1 într-un mod dependent de doză şi de concentraţia plasmatică. Concentraţiile plasmatice de aprepitant obţinute după o schemă terapeutică de 3 zile cu EMEND la adulți furnizează o saturare de peste 95 % a receptorilor cerebrali NK1.
5.3 Date preclinice de siguranţă
Datele preclinice nu au evidenţiat niciun risc special pentru om , pe baza studiilor convenţionale de toxicitate după administrarea unică și după doze repetate, genotoxicitate, potențial carcinogen, de toxicitate asupra funcţiei de reproducere şi dezvoltării. Cu toate acestea, trebuie reţinut că expunerea sistemică la rozătoare a fost similară sau chiar inferioară expunerii terapeutice la om la doza de 125 mg/80 mg. În particular, deşi în studiile asupra funcţiei reproductive nu s-au înregistrat reacţii adverse la nivelele de expunere umană, expunerile la animale nu sunt suficiente pentru a evalua în mod adecvat riscurile potenţiale la om.
Într-un studiu privind toxicitatea juvenilă la şobolanii trataţi din ziua postnatală 10 şi până în ziua 63, aprepitantul a condus la o deschidere vaginală timpurie la femele, începând cu doza de 250 mg/kg de două ori pe zi şi la o separare prepuţială întârziată la masculi, începând cu doză de 10 mg/kg de două ori pe zi. Nu au existat limite privind expunerea relevantă clinic. Nu s-au înregistrat efecte legate de tratament asupra împerecherii, fertilității sau privind supravieţuirea embrionară/fetală şi nu s-au produs modificări patologice la nivelul organelor reproducătoare. Într-un studiu privind toxicitatea juvenilă la câinii trataţi din ziua postnatală 14 până la ziua 42, s-a observat reducerea greutății testiculare şi a dimensiunii celulelor Leydig la masculi la doza de 6 mg/kg/zi şi creșterea greutății uterului, hipertrofia uterului şi a colului uterin şi edemul ţesutului vaginal au fost observate la femele începând cu doza de 4 mg/kg/zi. Nu au existat limite privind expunerea relevantă clinic la aprepitant. În cazul tratamentului pe termen scurt confirm schemei terapeutice recomandate ale dozelor, aceste rezultate nu sunt considerate a fi relevante clinic.
6. PROPRIETĂŢI FARMACEUTICE
6.1 Lista excipienţilor
Hidroxipropilceluloză (E 463)
Laurilsulfat de sodiu
Zahăr
Lactoză (anhidră)
Oxid roşu de fer (E 172)
Stearil fumarat de sodiu
6.2 Incompatibilităţi
Nu este cazul.
6.3 Perioada de valabilitate
Plic nedeschis: 2 ani
După reconstituire: Suspensia orală poate fi menținută la temperatura camerei (nu peste 30ºC) până la 3 ore. De asemenea poate fi păstrată la frigider (între 2ºC și 8ºC) până la 72 ore.
6.4 Precauţii speciale pentru păstrare
Acest medicament nu necesită condiţii de temperatură speciale de păstrare. A se păstra în ambalajul original pentru a fi protejat de umiditate.
Pentru condiţiile de păstrare ale medicamentului după reconstituire, vezi pct. 6.3.
6.5 Natura şi conţinutul ambalajului
Plicuri din PET/Al/PEJDL.
Cutie pentru o singură utilizare.
Fiecare cutie conţine un plic de pulbere pentru suspensie orală, o seringă dozatoare pentru administrare orală de 1 ml și una de 5 ml (polipropilenă cu garnitură inelară din silicon), un capac şi un pahar de amestec (polipropilenă).
6.6 Precauţii speciale pentru eliminarea reziduurilor şi alte instrucţiuni de manipulare
Conținutul fiecărui plic pentru o singură utilizare se folosește pentru a pregăti o suspensie cu 4,6 ml de apă, obţinând o concentraţie finală de 25 mg/ml.
- Pentru detalii suplimentare privind prepararea şi administrarea suspensiei, consultaţi prospectul și instrucţiunile de preparare a suspensiei orale pentru profesioniștii din domeniul sănătății.
- Utilizaţi seringa dozatoare de 5 ml pentru administrare orală pentru a măsura cei 4,6 ml de apă care se adaugă în paharul de amestec.
- Turnaţi tot conţinutul plicului în cei 4,6 ml de apă şi amestecaţi.
- După amestecare, măsuraţi volumul recomandat (doza) de suspensie cu ajutorul seringii dozatoare pentru administrare orală. Alegeți seringa dozatoare pentru administrare orală în funcție de doză. Utilizați seringa dozatoare de 1 ml pentru administare orală dacă doza este de 1 ml sau mai puțin și seringa dozatoare de 5 ml pentru administare orală dacă doza este mai mare de 1 ml. Administraţi doza pe cale orală. Dacă doza nu se administrează imediat după măsurare, seringa dozatoare umplută pentru administrare orală poate fi păstrată la frigider (între 2ºC și 8ºC) până la 72 ore înainte de utilizare.
- Înainte de administrare, suspensia orală poate fi păstrată la temperatura camerei (nu peste 30ºC) până la 3 ore.
- Aruncaţi suspensia rămasă şi materialele reziduale.
Orice medicament neutilizat sau material rezidual trebuie eliminat în conformitate cu reglementările locale.
7. DEŢINĂTORUL AUTORIZAŢIEI DE PUNERE PE PIAŢĂ
Merck Sharp & Dohme B.V.
Waarderweg 39
2031 BN Haarlem
Olanda
8. NUMĂRUL(ELE) AUTORIZAŢIEI DE PUNERE PE PIAŢĂ
EU/1/03/262/011
9. DATA PRIMEI AUTORIZĂRI SAU A REÎNNOIRII AUTORIZAŢIEI
Data primei autorizări: 11 noiembrie 2003
Data ultimei reînnoiri a autorizaţiei: 22 septembrie 2008
10. DATA REVIZUIRII TEXTULUI
Informaţii detaliate privind acest medicament sunt disponibile pe site-ul Agenţiei Europene pentru Medicamente http://www.ema.europa.eu.