MYNZEPLI 40 mg/ml
Rezumatul caracteristicilor produsului (RCP)
- 1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI
- 2. COMPOZIȚIA CALITATIVĂ ȘI CANTITATIVĂ
- 3. FORMA FARMACEUTICĂ
-
4. DATE CLINICE
- 4.1 Indicații terapeutice
- 4.2 Doze și mod de administrare
- 4.3 Contraindicații
- 4.4 Atenționări și precauții speciale pentru utilizare
- 4.5 Interacțiuni cu alte medicamente și alte forme de interacțiune
- 4.6 Fertilitatea, sarcina și alăptarea
- 4.7 Efecte asupra capacității de a conduce vehicule și de a folosi utilaje
- 4.8 Reacții adverse
- 4.9 Supradozaj
- 5. PROPRIETĂȚI FARMACOLOGICE
- 6. PROPRIETĂȚI FARMACEUTICE
- 7. DEȚINĂTORUL AUTORIZAȚIEI DE PUNERE PE PIAȚĂ
- 8. NUMĂRUL(ELE) AUTORIZAȚIEI DE PUNERE PE PIAȚĂ
- 9. DATA PRIMEI AUTORIZĂRI SAU A REÎNNOIRII AUTORIZAȚIEI
- 10. DATA REVIZUIRII TEXTULUI
- 1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI
- 2. COMPOZIȚIA CALITATIVĂ ȘI CANTITATIVĂ
- 3. FORMA FARMACEUTICĂ
-
4. DATE CLINICE
- 4.1 Indicații terapeutice
- 4.2 Doze și mod de administrare
- 4.3 Contraindicații
- 4.4 Atenționări și precauții speciale pentru utilizare
- 4.5 Interacțiuni cu alte medicamente și alte forme de interacțiune
- 4.6 Fertilitatea, sarcina și alăptarea
- 4.7 Efecte asupra capacității de a conduce vehicule și de a folosi utilaje
- 4.8 Reacții adverse
- 4.9 Supradozaj
- 5. PROPRIETĂȚI FARMACOLOGICE
- 6. PROPRIETĂȚI FARMACEUTICE
- 7. DEȚINĂTORUL AUTORIZAȚIEI DE PUNERE PE PIAȚĂ
- 8. NUMĂRUL(ELE) AUTORIZAȚIEI DE PUNERE PE PIAȚĂ
- 9. DATA PRIMEI AUTORIZĂRI SAU A REÎNNOIRII AUTORIZAȚIEI
- 10. DATA REVIZUIRII TEXTULUI
1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI
MYNZEPLI 40 mg/ml soluție injectabilă în seringă preumplută.
2. COMPOZIȚIA CALITATIVĂ ȘI CANTITATIVĂ
1 ml soluție injectabilă conține aflibercept* 40 mg.
O seringă preumplută conține un volum extractibil de cel puțin 0,09 ml, echivalent cu aflibercept cel puțin 3,6 mg. Acesta furnizează o cantitate utilizabilă pentru administrarea unei doze unice de 0,05 ml, conținând aflibercept 2 mg pentru pacienți adulți.
*Proteina de fuziune este formată din fragmente VEGF uman (factor endotelial de creștere vasculară) din domeniile extracelulare ale receptorilor 1 și 2, fuzionate cu fragmentul Fc a IgG1 uman și obținută prin tehnologie ADN recombinantă, în celule ovariene de hamster chinezesc (CHO) K1.
Pentru lista tuturor excipienților, vezi pct. 6.1.
3. FORMA FARMACEUTICĂ
Soluție injectabilă (injecție)
Soluția este limpede, incoloră până la galben pal și izoosmotică.
4. DATE CLINICE
4.1 Indicații terapeutice
MYNZEPLI este indicat la adulți pentru tratamentul:
- degenerescenței maculare legată de vârstă (DMLV) forma neovasculară (umedă) (vezi pct. 5.1),
- afectării acuității vizuale determinată de edemul macular secundar ocluziei venei retinei (OVR de ram sau OVR centrală) (vezi pct. 5.1),
- afectării acuității vizuale determinată de edemul macular diabetic (EMD) (vezi pct. 5.1),
- afectării acuității vizuale determinată de neovascularizația coroidală miopică (NVC miopică) (vezi pct. 5.1).
4.2 Doze și mod de administrare
MYNZEPLI se administrează numai sub formă de injecții intravitreene.
MYNZEPLI trebuie administrat numai de către un medic oftalmolog cu experiență în administrarea injecțiilor intravitreene.
Doze
DMLV forma umedă
Doza recomandată de MYNZEPLI este de 2 mg aflibercept, echivalent cu 0,05 ml.
Tratamentul cu MYNZEPLI este inițiat cu o injecție o dată pe lună, pentru trei administrări consecutive. Intervalul de tratament este apoi prelungit la două luni.
Pe baza interpretării de către medic a rezultatelor funcției vizuale și/sau modificărilor anatomice, intervalul de tratament poate fi menținut la două luni sau extins suplimentar, cu un regim de dozaj de tip „tratament și extindere”, crescând intervalele de injectare în incrementuri de 2 sau 4 săptămâni, astfel încât rezultatele vizuale și/sau anatomice să fie menținute stabile. În cazul în care rezultatele vizuale și/sau anatomice se deteriorează, intervalul de administrare a tratamentului trebuie scăzut în mod corespunzător.
Nu există nicio cerință pentru monitorizarea între injectări. În funcție de evaluarea medicului, programul vizitelor de monitorizare poate avea o frecvență mai mare decât cel al vizitelor pentru injectare.
Nu au fost studiate intervale de tratament între injectări mai mari de patru luni sau mai mici de patru săptămâni (vezi pct. 5.1).
Edem macular secundar OVR (OVR de ram sau OVR centrală)
Doza recomandată de MYNZEPLI este de 2 mg aflibercept, echivalent cu 0,05 ml.
După injectarea inițială, tratamentul este administrat lunar. Intervalul dintre 2 doze nu trebuie să fie mai mic de o lună.
În cazul în care rezultatele vizuale și anatomice indică faptul că pacientul nu prezintă beneficii în cadrul tratamentului continuu, MYNZEPLI trebuie întrerupt.
Tratamentul lunar continuă până când se obține acuitatea vizuală maximă și/sau nu există semne de activitate a bolii. Poate fi necesară administrarea lunară, timp de trei luni consecutiv, sau mai mult.
Tratamentul poate fi continuat cu un regim de tip „tratament și extindere“, crescând progresiv intervalele de administrare a tratamentului, astfel încât rezultatele vizuale și/sau anatomice să fie menținute stabile, însă nu există date suficiente pentru a concluziona referitor la durata acestui interval. În cazul în care rezultatele vizuale și/sau anatomice se deteriorează, intervalul de administrare a tratamentului trebuie scăzut în mod corespunzător.
Schema de monitorizare și tratament trebuie stabilită de către medicul curant în funcție de răspunsul individual al pacientului.
Monitorizarea activității bolii poate include examen clinic, teste funcționale sau tehnici imagistice (ex. tomografie în coerență optică sau angiofluorografie).
Edem macular diabetic
Doza recomandată de MYNZEPLI este de 2 mg aflibercept, echivalent cu 0,05 ml.
Tratamentul cu MYNZEPLI este început cu o injecție o dată pe lună pentru cinci administrări consecutive, urmat de o injecție la interval de două luni.
Pe baza interpretării de către medic a rezultatelor funcției vizuale și/sau modificărilor anatomice, intervalul de tratament poate fi menținut la 2 luni sau individualizat, cu un regim de tip „tratament și extindere”, crescând de regulă intervalul de administrare a tratamentului cu incrementuri de 2 săptămâni, astfel încât rezultatele funcției vizuale și/sau anatomice să fie menținute stabile. Există date limitate pentru intervale de tratament mai mari de 4 luni. În cazul în care rezultatele vizuale și/sau anatomice se deteriorează, intervalul de administrare a tratamentului trebuie scăzut în mod corespunzător.
Intervalele de tratament mai scurte de 4 săptămâni nu au fost studiate (vezi pct. 5.1).
Programul de monitorizare trebuie să fie stabilit de către medicul curant.
Dacă rezultatele vizuale și anatomice indică faptul că pacientul nu prezintă beneficii prin continuarea tratamentului, administrarea MYNZEPLI trebuie oprită.
Neovascularizația coroidală miopică
Doza recomandată de MYNZEPLI este de o singură injecție intravitreană de aflibercept 2 mg, echivalent cu 0,05 ml.
Pot fi administrate doze suplimentare dacă rezultatele vizuale și/sau anatomice indică faptul că boala persistă. Recurențele trebuie tratate drept o nouă manifestare a bolii.
Programul de monitorizare trebuie stabilit de către medicul curant.
Intervalul dintre două doze nu trebuie să fie mai scurt de o lună.
Grupe speciale de pacienți
Insuficiență hepatică și/sau renală
Nu s-au efectuat studii specifice cu MYNZEPLI la pacienți cu insuficiență hepatică și/sau renală.
Datele disponibile nu sugerează necesitatea ajustării dozei de MYNZEPLI la acești pacienți (vezi pct. 5.2).
Vârstnici
Nu sunt necesare precauții speciale. Există experiență limitată privind utilizarea la pacienți cu vârsta peste 75 ani cu EMD.
Copii și adolescenți
Siguranța și eficacitatea MYNZEPLI la copii și adolescenți cu vârsta sub 18 ani. Nu există date relevante pentru utilizarea afliberceptului la copii și adolescenți pentru indicațiile DMLV forma umedă, OVCR, ORVR, EMD și NVC miopică.
Mod de administrare
Injecțiile intravitreene trebuie efectuate de către un medic oftalmolog cu experiență în administrarea injecțiilor intravitreene, conform standardelor medicale și ghidurilor în vigoare. În general, trebuie să se asigure condiții adecvate de anestezie și asepsie, inclusiv administrarea locală a unui bactericid cu spectru larg (de exemplu, povidonă iodată aplicată la nivelul pielii perioculare, pleoapei și suprafeței oculare). Se recomandă dezinfecția chirurgicală a mâinilor, utilizarea mănușilor sterile, a unor câmpuri sterile și a unui specul de pleoape steril (sau echivalent).
Imediat după injectarea intreavitreană, pacienții trebuie monitorizați pentru creșterea presiunii intraoculare. Monitorizarea adecvată poate consta în verificarea perfuzării nervului optic sau tonometrie. Dacă este necesar, trebuie să fie disponibil echipament steril pentru paracenteza camerei anterioare.
După injectarea intravitreană, pacienții adulți trebuie instruiți să raporteze fără întârziere orice simptome sugestive de endoftalmită (de exemplu, durere oculară, înroșirea ochiului, fotofobie, vedere încețoșată).
Fiecare seringă preumplută trebuie utilizată numai pentru tratamentul unui singur ochi. Extragerea dozelor multiple dintr-o singură seringă preumplută poate crește riscul de contaminare și ulterior, infecție.
Seringa preumplută conține mai mult decât doza recomandată de aflibercept 2 mg (echivalent cu 0,05 ml soluție injectabilă). Volumul extractibil dintr-o seringă este cantitatea care poate fi eliminată din seringă și nu se foloseṣte în totalitate. Pentru MYNZEPLI seringă preumplută, volumul extractibil este de cel puțin 0,09 ml. Volumul în exces trebuie eliminat înainte de injectarea dozei recomandate (vezi pct. 6.6).
Injectarea întregului volum al seringii preumplute poate duce la supradozaj. Pentru a elimina bulele de aer din seringă împreună cu volumul în exces de medicament, se va împinge pistonul astfel încât să se alinieze baza cupolei acestuia (nu vârful cupolei) cu linia de marcare a dozei de pe seringă (echivalent cu 0,05 ml, ce conțin 2 mg aflibercept) (vezi pct. 4.9 și 6.6).
Acul pentru injectare trebuie introdus la 3,5-4,0 mm în spatele limbului, în cavitatea vitroasă, evitându-se meridianul orizontal în direcția centrului globului ocular. Apoi se administrează volumul injectabil de 0,05 ml; pentru injecțiile ulterioare trebuie să se utilizeze o zonă sclerală diferită.
După injectare, orice medicament neutilizat trebuie eliminat.
Pentru manipularea medicamentului înainte de administrare, vezi pct. 6.6.
4.3 Contraindicații
Hipersensibilitate la substanța activă sau la oricare dintre excipienții enumerați la pct. 6.1.
Infecție oculară sau perioculară activă sau suspectată.
Inflamație intraoculară activă, severă.
4.4 Atenționări și precauții speciale pentru utilizare
Trasabilitate
Pentru a avea sub control trasabilitatea medicamentelor biologice, numele și numărul lotului medicamentului administrat trebuie înregistrate cu atenție.
Reacții asociate injectării intravitreene
Injecțiile intravitreene, inclusiv cele cu MYNZEPLI, au fost asociate cu endoftalmită, inflamație intraoculară, dezlipire regmatogenă de retină, ruptură de retină și cataractă traumatică iatrogenă (vezi pct. 4.8). Atunci când se administrează MYNZEPLI, trebuie utilizate întotdeauna tehnici de injectare aseptice adecvate. În plus, pacienții trebuie monitorizați în timpul săptămânii după injectare, pentru a permite tratamentul precoce în cazul apariției unei infecții.
Pacienții adulți trebuie instruiți să raporteze fără întârziere orice simptom care sugerează o endoftalmită sau oricare dintre evenimentele menționate mai sus.
Seringa preumplută conține mai mult decât doza recomandată de aflibercept 2 mg (echivalentă cu 0,05 ml) pentru pacienți adulți. Volumul în exces trebuie eliminat înainte de administrare (vezi pct. 4.2 și 6.6). S-au observat creșteri ale presiunii intraoculare în decurs de 60 minute de la administrarea unei injecții intravitreene, inclusiv la cele cu MYNZEPLI (vezi pct. 4.8). Sunt necesare precauții speciale la pacienții cu glaucom insuficient controlat prin tratament (nu se injectează MYNZEPLI dacă presiunea intraoculară este ≥ 30 mmHg). În toate cazurile, atât presiunea intraoculară cât și perfuzia la nivelul rădăcinii nervului optic trebuie monitorizate și tratate corespunzător.
Imunogenitate
Deoarece este o proteină folosită în scop terapeutic există potențial de imunogenitate în cazul administrării afliberceptului (vezi pct. 4.8). Pacienții trebuie instruiți să raporteze orice semn sau simptom de inflamație intraoculară, de exemplu durere, fotofobie sau roṣeață, care ar putea fi semne clinice atribuite hipersensibilității.
Efecte sistemice
Reacțiile adverse sistemice includ hemoragii, altele decât cele oculare, și evenimente tromboembolice arteriale care au fost raportate după administrarea intravitreană a inhibitorilor VEGF și există un risc teoretic ca acestea să fie legate de inhibarea VEGF. Există date limitate privind siguranța tratamentului la pacienții cu OVCR, ORVR, EMD sau NVC miopică cu antecedente de accident vascular cerebral sau accidente ischemice tranzitorii sau infarct miocardic în ultimele 6 luni. Trebuie exercitată precauție în cazul în care sunt tratați acești pacienți.
Alte informații:
Similar altor tratamente intravitreene anti-VEGF pentru DMLV forma umedă, OVCR, ORVR, EMD și NVC miopică, următoarele informații sunt, de asemenea, valabile:
- Siguranța și eficacitatea tratamentului cu aflibercept administrat concomitent la ambii ochi nu au fost studiate în mod sistematic (vezi pct. 5.1). Dacă tratamenul bilateral este efectuat în același timp, acest lucru poate duce la o expunere sistemică crescută, ceea ce poate crește riscul reacțiilor adverse sistemice.
- Utilizarea concomitentă a altor anti-VEGF (factorul de creștere al endoteliului vascular) Nu există date disponibile referitoare la utilizarea concomitentă a afliberceptului cu alte medicamente anti-VEGF (sistemice sau oculare).
- Factorii de risc asociați cu apariția unei rupturi la nivelul epiteliului pigmentar al retinei după tratamentul anti-VEGF pentru DMLV forma umedă includ detașarea mare și/sau profundă a epiteliului pigmentar al retinei. Inițierea tratamentului cu aflibercept trebuie efectuată cu precauție la pacienții care prezintă acești factori de risc privind rupturile epiteliului pigmentar al retinei.
- Tratamentul trebuie întrerupt la pacienții cu dezlipire regmatogenă de retină sau cu perforații maculare de stadiul 3 sau 4.
- În cazul rupturii de retină tratamentul trebuie întrerupt și nu trebuie reluat până la vindecarea țesutului epitelial al retinei.
- Tratamentul trebuie întrerupt și nu trebuie reluat mai devreme de momentul în care este programată administrarea următoarei doze, în cazul:
- scăderii celei mai bune acuități vizuale corectate (BAVC) cu ≥ 30 litere comparativ cu ultima verificare a acuității vizuale
- unei hemoragii subretiniene care a inclus centrul foveei sau dacă suprafața hemoragiei este ≥ 50 % din aria lezată totală.
- Tratamentul trebuie întrerupt și nu trebuie reluat mai devreme de 28 zile înainte sau după planificarea sau efectuarea unei intervenții chirurgicale intraoculare.
- MYNZEPLI nu trebuie utilizat în timpul sarcinii, cu excepția cazului în care beneficiile potențiale depășesc riscul potențial pentru făt (vezi pct. 4.6).
- Femeile aflate la vârsta fertilă trebuie să utilizeze măsuri contraceptive eficace în timpul tratamentului și timp de cel puțin 3 luni după ultima injecție intravitreană cu aflibercept (vezi pct. 4.6).
- Experiența clinică în tratamentul pacienților cu OVCR și ORVR ischemic este limitată.Tratamentul nu este recomandat la pacienții cu semne clinice de pierdere ireversibilă ischemică a funcției vizuale.
Grupe de pacienți pentru care există date limitate
Există doar o experiență limitată în ceea ce privește tratamentul pacienților cu EMD determinat de diabetul zaharat de tip I sau al pacienților diabetici cu o valoare a HbA1c peste 12% sau cu retinopatie diabetică proliferativă.
Aflibercept nu a fost studiat la pacienții cu infecții sistemice active sau la pacienții cu afecțiuni concomitente oculare, cum ar fi dezlipirea de retină sau gaura maculară. De asemenea, nu există experiență în ceea ce privește tratamentul cu aflibercept la pacienții diabetici cu hipertensiune arterială necontrolată. Această lipsă a informațiilor trebuie luată în considerare de către medic atunci când tratează acești pacienți.
Pentru NVC miopică, nu există experiență privind utilizarea afliberceptului în tratamentul pacienților care nu aparțin rasei galbene, al pacienților la care s-a efectuat anterior tratament pentru NVC miopică și al pacienților cu leziuni extrafoveale.
Informații referitoare la excipienți
Acest medicament conține sodiu mai puțin de 1 mmol (23 mg) per doză, adică practic „nu conține sodiu”
4.5 Interacțiuni cu alte medicamente și alte forme de interacțiune
Nu s-au efectuat studii privind interacțiunile.
Nu s-a studiat utilizarea suplimentară a tratamentului fotodinamic (TFD) cu verteporfină și aflibercept, deci un profil de siguranță nu este stabilit încă.
Copii și adolescenți
Nu s-au efectuat studii privind interacțiunile.
4.6 Fertilitatea, sarcina și alăptarea
Femei aflate la vârsta fertilă
Femeile aflate la vârsta fertilă trebuie să utilizeze măsuri contraceptive eficace în timpul tratamentului și timp de cel puțin 3 luni după ultima injecție intravitreană cu aflibercept (vezi pct. 4.4).
Sarcina
Nu există date privind utilizarea aflibercept la femeile gravide. Studiile la animale au evidențiat toxicitate embriofetală (vezi pct. 5.3).
Cu toate că expunerea sistemică după administrarea oculară este foarte scăzută, MYNZEPLI nu trebuie utilizat în timpul sarcinii, cu excepția cazului în care beneficiile potențiale depășesc riscul potențial pentru făt.
Alăptarea
Pe baza unor date foarte limitate la om, aflibercept poate fi excretat în laptele uman la niveluri scăzute. Aflibercept este o moleculă proteică mare și cantitatea de medicament absorbită de copil este de așteptat să fie minimă. Efectele aflibercept asupra nou-născutului/sugarului alăptat sunt necunoscute.
Ca o măsură de precauție, alăptarea nu este recomandată în timpul utilizării MYNZEPLI.
Fertilitatea
Rezultatele provenite din studiile la animale, constând în expunere sistemică crescută, indică faptul că aflibercept poate afecta fertilitatea masculină și feminină (vezi pct. 5.3). Nu se anticipează asemenea efecte după administrarea oculară și expunere sistemică foarte scăzută.
4.7 Efecte asupra capacității de a conduce vehicule și de a folosi utilaje
Injecția cu MYNZEPLI are o influență minoră asupra capacității de a conduce vehicule și de a folosi utilaje din cauza tulburărilor vizuale temporare asociate cu administrarea injecțiilor sau cu examinarea oculară. Pacienții nu trebuie să conducă vehicule sau să folosească utilaje până când funcția lor vizuală nu s-a restabilit suficient.
4.8 Reacții adverse
Rezumatul profilului de siguranță
Populația de siguranță a fost constituită din 3102 pacienți, în opt studii clinice de fază III. Dintre aceștia, 2501 pacienți au fost tratați cu doza recomandată de 2 mg.
Reacțiile adverse oculare grave la nivelul ochiului evaluat în cadrul studiului, asociate cu procedura de injectare au apărut la mai puțin de 1 din 1900 injectări intravitreene cu aflibercept și au inclus: cecitate, endoftalmită, dezlipire de retină, cataractă traumatică, cataractă, hemoragie vitroasă, dezlipire de corp vitros și creștere a presiunii intraoculare (vezi pct. 4.4).
Reacțiile adverse observate cel mai frecvent (apărute la cel puțin 5% dintre pacienții cărora li s-a administrat aflibercept) au fost: hemoragie conjunctivală (25%), hemoragie retiniană (11%), scădere a acuității vizuale (11%), durere oculară (10%), cataractă (8%), creștere a presiunii intraoculare (8%), dezlipire de corp vitros (7%) și flocoane intravitreene (7%).
Rezumatul reacțiilor adverse – lista sub formă de tabel
Datele privind siguranța, descrise mai jos, includ toate reacțiile adverse provenite din cele opt studii clinice de fază III, în indicațiile DMLV forma umedă, OVCR, ORVR, EMD și NVC miopică, cu o posibilitate rezonabilă de cauzalitate legată de procedura de injectare sau de medicament.
Reacțiile adverse sunt enumerate în funcție de clasificarea pe aparate, sisteme și organe, utilizând următoarea convenție:
Foarte frecvente (≥1/10), frecvente (≥1/100 și <1/10), mai puțin frecvente (≥1/1000 și <1/100), rare (≥1/10000 și <1/1000), cu frecvență necunoscută (care nu poate fi estimată din datele disponibile).
În cadrul fiecărei grupe de frecvență, reacțiile adverse la medicament sunt prezentate în ordinea descrescătoare a gravității.
Tabelul 1: Toate reacțiile adverse la medicament asociate tratamentului, raportate la pacienți în studii clinice de fază III (date cumulate din studiile clinice de fază III pentru indicațiile DMLV forma umedă, OVCR, ORVR, EMD și NVC miopică) sau în timpul supravegherii după punerea pe piață
| Clasificarea pe aparate, sisteme și organe | Frecvența | Reacție adversă |
|---|---|---|
| Tulburări ale sistemului imunitar | Mai puțin frecvente | Hipersensibilitate*** |
| Tulburări oculare | Foarte frecvente | Scădere a acuității vizuale, Hemoragie retiniană, Hemoragie conjunctivală, Durere oculară |
| Frecvente | Ruptură a epiteliului retinian pigmentar*, Dezlipire a epiteliului retinian pigmentar, Degenerare retiniană, Hemoragie vitroasă, Cataractă, Cataractă corticală, Cataractă nucleară, Cataractă subcapsulară, Eroziune corneeană, Abraziune corneană, Creştere a presiunii intraoculare, Vedere înceţoşată, Flocoane intravitroase, Dezlipire de corp vitros, Durere la locul de injecţie, Senzaţie de corpi străini la nivel ocular, Hiperlacrimaţie, Edem palpebral, Hemoragie la locul de injecţie, Keratită punctată, Hiperemie conjunctivală, Hiperemie oculară. | |
| Mai puțin frecvente | Endoftalmită**, Dezlipire de retină, Ruptură de retină, Irită, Uveită, Iridocciclită, Opacități | |
| lenticulare, Defecte ale epiteliului cornean, Iritație la locul de injecție, Senzație anormală în ochi, Blefarită, Congestie la nivelul camerei anterioare, Edem cornean | ||
| Rare | Cecitate, Cataractă traumatică, Vitrită, Hipopion | |
| Cu frecvență necunoscută | Sclerită**** |
* Tulburări cunoscute a fi asociate cu DMLV forma umedă. Observate numai în studiile efectuate la pacienții cu DMLV forma umedă. Tulburări cunoscute a fi asociate cu DMLV forma umedă. Observate numai în studiile efectuate la pacienții cu DMLV forma umedă.
** Endoftalmite de cultură pozitivă și cultură negativă
*** În timpul perioadei de după punerea pe piață, raportările de hipersensibilitate au inclus erupții cutanate tranzitorii, prurit, urticarie, și cazuri izolate de reacții anafilactice severe/reacții anafilactoide. **** Din raportările post-marketing.
Descrierea reacțiilor adverse selectate
Într-un studiu clinic de fază III efectuat la pacienții cu DMLV forma umedă a fost observată o incidență crescută a hemoragiilor conjunctivale la pacienții tratați cu medicamente antiagregante. Această incidență crescută a fost comparabilă cu cea apărută la pacienții tratați cu ranibizumab și aflibercept.
Evenimentele arteriale tromboembolice (EAT) sunt evenimente adverse potențial corelate cu inhibiția sistemică a VEGF. Există un risc teoretic de evenimente arteriale tromboembolice, inclusiv accident vascular cerebral și infarct miocardic, în urma administrării intravitreene de inhibitori ai VEGF.
A fost observată o rată scăzută a incidenței evenimentelor tromboembolice arteriale în studiile clinice cu aflibercept la pacienții cu DMLV, EMD, OVR, NVC miopică și RP. Referitor la toate indicațiile, nu a fost observată nicio diferență notabilă între grupurile tratate cu aflibercept și grupurile comparator respective.
Similar tuturor proteinelor terapeutice, există un risc potențial de imunogenitate în cazul administrării afliberceptului.
Raportarea reacțiilor adverse suspectate
Raportarea reacțiilor adverse suspectate după autorizarea medicamentului este importantă. Acest lucru permite monitorizarea continuă a raportului beneficiu/risc al medicamentului. Profesioniștii din domeniul sănătății sunt rugați să raporteze orice reacție adversă suspectată prin intermediul sistemului național de raportare, astfel cum este menționat în Anexa V.
4.9 Supradozaj
În studiile clinice, s-au utilizat doze de până la 4 mg la intervale lunare și au apărut cazuri izolate de supradozaj la doze de 8 mg.
Supradozajul cu un volum crescut de soluție injectabilă poate crește presiunea intraoculară. Prin urmare, în cazul supradozajului trebuie monitorizată presiunea intraoculară și trebuie inițiat tratamentul adecvat de către medicul curant, dacă acest lucru este considerat necesar (vezi pct. 6.6).
5. PROPRIETĂȚI FARMACOLOGICE
5.1 Proprietăți farmacodinamice
Grupa farmacoterapeutică: Medicamente oftalmologice / Medicamente antineovascularizație Codul ATC: S01LA05
MYNZEPLI este un medicament biosimilar. . Informații detaliate sunt disponibile pe site-ul Agenției Europene pentru Medicamente https://www.ema.europa.eu
Aflibercept este o proteină recombinantă de fuziune, formată din porțiuni ale domeniilor extracelulare ale receptorilor 1 și 2 ai VEGF uman, fuzionate cu porțiunea Fc a IgG1 umane.
Aflibercept este obținut prin tehnologie ADN recombinantă în celule ovariene de hamster chinezesc (CHO) K1.
Aflibercept acționează ca un receptor capcană, solubil, care se leagă de VEGF-A și de factorul placentar de creștere (PlGF), cu afinitate superioară receptorilor naturali ai acestora și, astfel, poate inhiba legarea și activarea acestor receptori înrudiți ai VEGF.
Mecanism de acțiune
Factorul A endotelial de creștere vasculară (VEGF-A) și factorul placentar de creștere (PlGF) sunt membri ai familiei VEGF a factorilor angiogenici, care pot acționa ca factori puternici mitogeni, chemotactici și de permeabilitate vasculară pentru celulele endoteliale. VEGF acționează prin intermediul a doi receptori ai tirozin kinazelor, VEGFR-1 și VEGFR-2, prezenți pe suprafața celulelor endoteliale. PlGF se leagă numai de VEGFR-1, prezent de asemenea pe suprafața leucocitelor. Activarea în exces a acestor receptori de către VEGF-A poate determina apariția neovascularizației patologice și creșterea permeabilității vasculare. În aceste procese, PlGF poate acționa sinergic cu VEGF-A și este cunoscut, de asemenea, ca promotor al infiltrației leucocitare și inflamației vasculare.
Efecte farmacodinamice
DMLV forma umedă
DMLV forma umedă se caracterizează prin apariția neovascularizației patologice coroidale (NVC). Scurgerea sângelui și lichidelor de la nivelul NVC poate provoca îngroșare retiniană sau edem retinian și/sau hemoragie sub-/intraretiniană, ceea ce duce la pierderea acuității vizuale.
La pacienții tratați cu aflibercept (o injecție administrată lunar, timp de trei luni consecutiv, urmată de o injecție la intervale de 2 luni), grosimea centrală a retinei [GCR] a scăzut curând după inițierea tratamentului și dimensiunea medie a leziunii NVC a scăzut, ceea ce confirmă rezultatele observate în cazul administrării ranibizumabului lunar în doză de 0,5 mg.
În studiul clinic VIEW1 s-au evidențiat scăderi medii ale GCR la tomografia în coerență optică (TCO) a scăderii medii ale îngroșării retinei (-130 și -129 microni în săptămâna 52 pentru grupele de studiu la care s-a administrat aflibercept 2 mg la intervale de 2 luni și, respectiv, ranibizumab 0,5 mg administrat lunar). De asemenea, la momentul săptămânii 52 în cadrul studiului clinic VIEW2, TCO a evidențiat scăderi medii ale GCR (-149 și -139 microni pentru grupele de studiu la care s-au administrat aflibercept 2 mg la intervale de 2 luni și, respectiv, ranibizumab 0,5 mg administrat lunar). Scăderea dimensiunii NVC și scăderea GCR au fost în general menținute în al doilea an al studiilor.
Studiul clinic ALTAIR, efectuat la pacienți japonezi, cu DMLV forma umedă, cărora nu li se administrase tratament anterior, arată rezultate similare cu ale studiilor VIEW, folosind 3 injectări lunare inițiale, de aflibercept 2 mg, urmate de o injectare după alte 2 luni, iar apoi continuat cu un regim de tip „tratament și extindere” cu intervale de tratament variabile (ajustări de 2 sau 4 săptămâni) până la un interval de maximum 16 săptămâni, conform criteriilor specificate anterior. În săptămâna 52, au existat scăderi medii ale grosimii centrale a retinei (GCR) la TCO, de -134,4 și -126,1 microni pentru grupul cu ajustare de 2 săptămâni și respectiv grupul cu ajustare de 4 săptămâni. Proporția pacienților fără fluid la TCO în săptămâna 52 a fost de 68,3% și 69,1% în grupurile cu ajustare de 2 și respectiv 4 săptămâni. În general, scăderea GCR s-a păstrat în ambele brațe ale studiului ALTAIR în al doilea an.
Studiul clinic ARIES a fost proiectat pentru explorarea non-inferiorității unui regim de doze de tipul tratament și extindere cu aflibercept 2 mg inițiat imediat după administrarea inițială a 3 injectări lunare și o injectare suplimentară după 2 luni versus un regim de doze de tipul tratament și extindere inițiat la un an după tratament. Pentru pacienții care aveau nevoie de doze mai frecvente decât Q8, cel puțin o dată pe parcursul studiului, GCR a rămas mărită, dar scăderea medie a GCR de la nivelul de bază la săptămâna 104 a fost -160,4 microni, similară cu cea a pacienților tratați la Q8 sau intervale mai puțin frecvente.
Edem macular secundar OVCR și ORVR
În OVCR și ORVR apare ischemia retiniană, aceasta semnalând eliberarea VEGF care, la rândul său, destabilizează joncțiunile compacte și stimulează proliferarea celulelor endoteliale. Reglarea ascendentă a VEGF este asociată cu ruperea barierei hemato-retiniene, permeabilitate vasculară crescută, edem retinian și complicații ale neovascularizației.
La pacienții tratați cu o injecție de aflibercept 2 mg lunar, timp de 6 luni consecutiv, s-a observat un răspuns consistent, rapid și intens în ceea ce privește morfologia (măsurată prin îmbunătățirea medie a GCR). În săptămâna 24, reducerea GCR a fost statistic superioară față de grupul de control, în toate cele 3 studii clinice (COPERNICUS în OVCR: -457 vs. -145 microni; GALILEO în OVCR: -449 vs. -169 microni; VIBRANT în ORVR: -280 vs. -128 microni). Această reducere a GCR față de momentul inițial s-a menținut până la sfârșitul fiecărui studiu clinic, săptămâna 100 în studiul clinic COPERNICUS, săptămâna 76 în studiul clinic GALILEO și săptămâna 52 în studiul clinic VIBRANT.
Edem macular diabetic
Edemul macular diabetic este o consecință a retinopatiei diabetice și se caracterizează prin creșterea permeabilității vasculare și deteriorarea capilarelor retiniene, care poate duce la pierderea acuității vizuale.
La pacienții tratați cu aflibercept, cea mai mare parte dintre aceștia având diagnostic de diabet zaharat de tip 2, s-a observat un răspuns rapid și intens în ceea ce privește morfologia (GCR, scorul DRSS).
În studiile clinice VIVIDDME și VISTADME, scăderile medii semnificativ statistic mai mari ale GCR față de valoarea inițială în săptămâna 52 au fost observate la pacienții tratați cu aflibercept comparativ cu pacienții din grupul de control cu laser, fiind de -192,4 microni și -183,1 microni pentru grupurile tratate cu aflibercept 2Q8 și, respectiv, -66,2 microni și -73,3 microni pentru grupurile de control. În săptămâna 100, scăderea s-a menținut, cu -195,8 microni și -191,1 microni pentru grupurile tratate cu aflibercept 2Q8 și respectiv cu -85,7 și -83,9 microni pentru grupurile de control, în studiile VIVIDDME și VISTADME.
În studiile VIVIDDME și VISTADME, o îmbunătățire ≥ 2 trepte a scorului DRSS (Scorul severității retinopatiei diabetice) a fost evaluată în manieră prespecificată. Scorul DRSS a putut fi clasificat pe grade la 73,7% dintre pacienții din studiul VIVIDDME și la 98,3% dintre pacienții din studiul VISTADME. În săptămâna 52, la 27,7% și 29,1% dintre pacienții din grupurile tratate cu aflibercept 2Q8 și la 7,5% și 14,3% dintre pacienții din grupurile de control s-a înregistrat o îmbunătățire ≥ 2 trepte a scorului DRSS. În săptămâna 100, procentele respective au fost de 32,6% și 37,1% dintre pacienții din grupurile tratate cu aflibercept 2Q8 și 8,2% și 15,6% dintre pacienții din grupurile de control.
Studiul clinic VIOLET a comparat trei regimuri diferite de dozare a aflibercept 2 mg pentru tratamentul EMD după cel puțin un an de tratament la intervale fixe, când tratamentul a fost inițiat cu 5 doze lunare consecutive urmate de doze la fiecare 2 luni. În săptămâna 52 și săptămâna 100 ale studiului, adică al doilea și al treilea an de tratament, media schimbărilor GCR era similară din punct de vedere clinic pentru “tratament și extindere” (2T&E), pro re nata (2PRN) și 2Q8, respectiv cu variații de -2,1, 2,2 și -18,8 microni în săptămâna 52 și variații de 2,3, -13,9 și -15,5 microni în săptămâna 100.
Neovascularizație coroidală miopică
Neovascularizația coroidală miopică (NVC miopică) este o cauză frecventă a pierderii vederii la adulții cu miopie patologică. Aceasta apare ca mecanism de vindecare a leziunilor, în urma ruperii membranei Bruch și reprezintă cel mai dăunător eveniment pentru vedere în miopia patologică.
La pacienții tratați cu aflibercept în studiul clinic MYRROR (o injecție administrată la începutul terapiei, fiind administrate injecții suplimentare în cazul persistenței sau al recurenței bolii), GCR s-a diminuat la scurt timp după inițierea tratamentului fiind în favoarea tratamentului cu aflibercept în săptămâna 24 (-79 microni pentru grupul tratat cu aflibercept 2 mg și, respectiv, -4 microni pentru grupul de control), care s-a menținut până în săptămâna 48. În plus, leziunea NVC medie s-a redus.
Eficacitate și siguranță clinică
DMLV forma umedă
Siguranța și eficacitatea clinică a afliberceptului au fost evaluate în două studii randomizate, multicentrice, cu dublă mascare a formei farmaceutice, controlate activ, la pacienți cu DMLV forma umedă (VIEW1 și VIEW2) cu un număr total de 2412 pacienți tratați și evaluați în vederea stabilirii eficacității (1817 pacienți la care s-a administrat aflibercept).Vârsta pacienților a variat de la 49 ani la 99 ani, cu o medie de 76 ani. În aceste studii clinice, aproximativ 89% (1,616/1,817) dintre pacienții randomizați la tratamentul cu aflibercept aveau vârsta de 65 ani sau mai mult, iar aproximativ 63% (1,139/1,817) aveau vârsta de 75 ani sau mai mult. În cadrul fiecărui studiu, pacienții au fost repartizați randomizat, în raport de 1:1:1:1, la unul din cele 4 regimuri de dozaj:
- aflibercept administrat în doză de 2 mg la intervale de 8 săptămâni, după administrarea a 3 doze inițiale la intervale lunare aflibercept 2Q8);
- aflibercept administrat în doză de 2 mg la intervale de 4 săptămâni (aflibercept 2Q4);
- aflibercept administrat în doză de 0,5 mg la intervale de 4 săptămâni (aflibercept 0,5Q4); și
- ranibizumab administrat în doză de 0,5 mg la intervale de 4 săptămâni (ranibizumab 0,5Q4).
În al doilea an al studiilor, pacienților li s-a administrat în continuare doza la care au fost inițial repartizați randomizat, dar cu un program modificat de administrare a dozelor, pe baza evaluării rezultatelor vizuale și anatomice, cu un interval maxim de dozaj de 12 săptămâni, definit de protocol.
În ambele studii, obiectivul principal al eficacității a fost procentul pacienților din grupele de studiu, care șiau menținut acuitatea vizuală, de exemplu pierderea a mai puțin de 15 litere din acuitatea vizuală în săptămâna 52, de la momentul inițial.
În studiul clinic VIEW1, în săptămâna 52, 95,1% dintre pacienții din grupul tratat cu aflibercept 2Q8 au menținut acuitatea vizuală, comparativ cu 94,4% dintre pacienții în grupul căruia s-a administrat ranibizumab 0,5Q4. În studiul clinic VIEW2, în săptămâna 52, 95,6% dintre pacienții din grupul tratat cu aflibercept 2Q8 și- au menținut acuitatea vizuală, comparativ cu 94,4% dintre pacienții în grupul căruia s-a administrat ranibizumab 0,5Q4. În ambele studii, tratamentul cu aflibercept s-a dovedit a fi non-inferior și echivalent din punct de vedere clinic cu grupul căruia s-a administrat ranibizumab 0,5Q4.
Rezultatele detaliate provenite din analiza centralizată a ambelor studii sunt prezentate în Tabelul 2 și Figura 1 de mai jos.
Tabelul 2: Rezultatele privind eficacitatea în săptămâna 52 (analiză primară) și în săptămâna 96; date combinate provenite din studiile VIEW1 și VIEW2B)
| Rezultat privind eficacitatea | Aflibercept 2Q8 E) (aflibercept 2 mg la intervale de 8 săptămâni, după administrarea a 3 doze inițiale la intervale lunare) (N = 607) | Ranibizumab 0,5Q4 (ranibizumab 0,5 mg la intervale de 4 săptămâni) (N = 595) | ||
| Săptămâna 52 | Săptămâna 96 | Săptămâna 52 | Săptămâna 96 | |
| Numărul mediu de injecții | 7,6 | 11,2 | 12,3 | 16,5 |
| Numărul mediu de injecții din săptămâna 52 până în săptămâna 96 | 4,2 | 4,7 | ||
Procentul pacienților cu < 15 litere pierdute față de momentul inițial SPPA) | 95,33%B) | 92,42% | 94,42%B) | 91,60% |
| DiferențăC) (IÎ95%)D) | 0,9% (-1,7; 3,5)F) | 0,8% (-2,3; 3,8)F) | ||
| Modificarea medie a AVOC măsurată prin scorul literelor ETDRSA) față de momentul inițial | 8,40 | 7,62 | 8,74 | 7,89 |
Diferența în media celor mai mici pătrate A) (litere ETDRS)C) (IÎ95%)D) | -0,32 (-1,87; 1,23) | -0,25 (-1,98; 1,49) | ||
Procentul pacienților cu ≥ 15 litere câștigate față de momentul inițial | 30,97% | 33,44% | 32,44% | 31,60% |
DiferențăC) IÎ(95%)D) | -1,5% (-6,8; 3,8) | 1,8% (-3,5; 7,1) | ||
A) AVOC: Acuitatea vizuală optim corectată
ETDRS: Early Treatment Diabetic Retinopathy Study (Studiul retinopatiei diabetice cu tratament precoce) LS: Media celor mai mici pătrate derivată din ANCOVA
SPP: Set Per Protocol (set pentru fiecare protocol)
B) Setul complet de analiză (FAS), Extrapolarea în sens longitudinal a ultimelor date observate (LOCF) pentru toate analizele, cu excepția procentului de pacienți la care acuitatea vizuală a fost menținută în săptămâna 52, reprezentând setul pentru fiecare protocol (PPS)
C) Diferența este reprezentată de valoarea grupului cu aflibercept minus valoarea grupului cu ranibizumab. O valoare pozitivă favorizează aflibercept.
D) Intervalul de încredere (IÎ) calculat prin aproximare normală
E) După inițierea tratamentului cu doze administrate la intervale de trei luni
F) Un interval de încredere complet superior valorii de -10% indică non-inferioritatea aflibercept față de ranibizumab
Figura 1. Modificarea medie a acuității vizuale de la momentul inițial până în săptămâna 96 pentru datele centralizate provenite din studiile View1 și View2
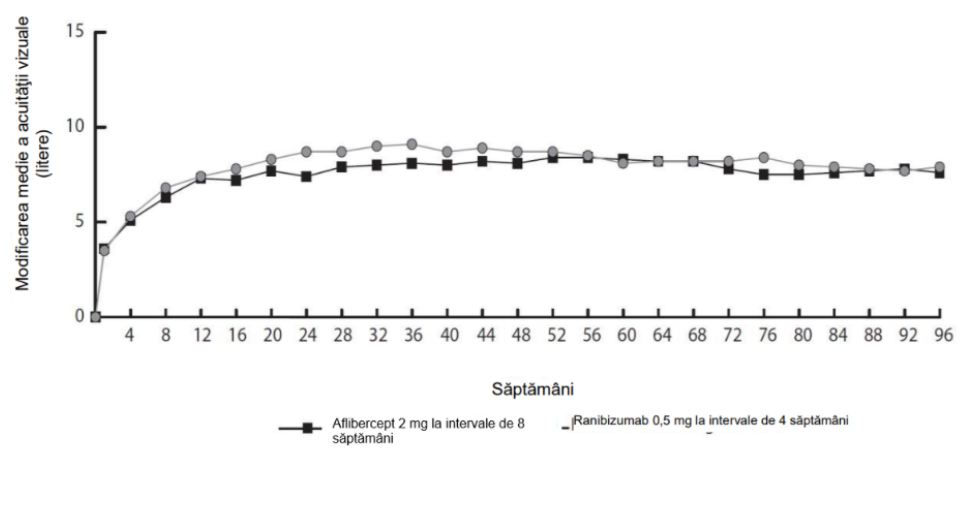
În analiza datelor centralizate provenite din studiile VIEW1 și VIEW2, aflibercept a demonstrat modificări semnificative din punct de vedere clinic față de momentul inițial, conform Chestionarului privind funcția vizuală, al Institutului Național pentru Afecțiuni Oculare (NEI VFQ-25), fără diferențe semnificative clinic față de ranibizumab. Importanța acestor modificări a fost similară celor observate în studiile publicate, corespunzând unui câștig de 15 litere din acuitatea vizuală optim corectată (AVOC).
În al doilea an al studiilor, eficacitatea s-a menținut, în general, conform ultimei evaluări din săptămâna 96, și 2-4% dintre pacienți au necesitat administrarea lunară a tuturor injecțiilor, iar 1/3 dintre pacienți au necesitat cel puțin o injecție la un interval de tratament de numai o lună.
Scăderile medii ale zonei NVC au fost evidente la toate grupele de tratament în ambele studii.
Rezultatele privind eficacitatea în toate subgrupele care au putut fi evaluate (de exemplu vârstă, sex, rasă, acuitate vizuală la momentul inițial, tipul leziunii, mărimea leziunii) în fiecare studiu și în analiza combinată au confirmat rezultatele în populația generală.
ALTAIR a fost un studiu multicentric de 96 de săptămâni, randomizat, deschis, efectuat la 247 pacienți japonezi cu DMLV forma umedă, naivi la tratament, dezvoltat pentru a evalua eficacitatea și siguranța afliberceptului după două intervale (2 săptămâni și 4 săptămâni) de ajustare a unui regim de tip „tratament și extindere”.
Tuturor pacienților li s-au administrat doze lunare de aflibercept de 2 mg, timp de 3 luni, urmate de o injectare după un interval de 2 luni. În săptămâna 16, pacienții au fost randomizați 1:1 în două grupuri de tratament: 1) aflibercept „tratament și extindere” cu ajustări la 2 săptămâni și 2) aflibercept „tratament și extindere” cu ajustări la 4 săptămâni. Decizia de a extinde sau a scurta intervalele de tratament a fost luată în funcție de criteriile vizuale și/sau anatomice definite de protocol, cu un interval de tratament de maximum 16 săptămâni pentru ambele grupuri.
Criteriul principal de evaluare a eficacității a fost schimbarea medie a AVOC de la nivelul de bază la săptămâna 52. Criteriile secundare de evaluare a eficacității au fost reprezentate de proporția de pacienți ce nu au pierdut ≥15 litere și proporția de pacienți care au câștigat cel puțin 15 litere din AVOC de la nivelul de bază la săptămâna 52.
În săptămâna 52, pacienții din brațul de „tratament și extindere” cu ajustări la 2 săptămâni au obținut un câștig mediu de 9,0 litere din nivelul de bază, comparativ cu 8,4 litere pentru cei din grupul cu ajustare la 4 săptămâni [diferența medie LS în litere (95% IÎ): -0,4 (-3,8, 3,0), ANCOVA]. Proporția de pacienți care nu au pierdut ≥15 litere în cele două brațe de tratament a fost similară (96,7% la grupul cu ajustare la 2 săptămâni și 95,9% la 4 săptămâni). Proporția de pacienți care au avut un câștig ≥15 litere în săptămâna 52 a fost 32,5% în grupul cu ajustare la 2 săptămâni și 30,9% în grupul cu ajustare la 4 săptămâni. Proporția de pacienți la care s-a extins intervalul de tratament la 12 săptămâni sau mai mult a fost 42,3% în grupul cu ajustare la 2 săptămâni și 49,6% în grupul cu ajustare la 4 săptămâni. Suplimentar, în grupul cu ajustare la 4 săptămâni, la 40,7% din pacienți s-a extins intervalul de tratament la 16 săptămâni. La ultima vizită până la săptămâna 52, 56,8% și 57,8% dintre pacienții din grupurile cu ajustare la 2 și respectiv 4 săptămâni au avut următoarea injectare programată la un interval de 12 săptămâni sau mai mare.
În al doilea an de studiu, în general eficacitatea s-a păstrat până la și incluzând ultima evaluare din săptămâna 96, cu un câștig mediu față de nivelul de bază de 7,6 litere în grupul cu ajustare la 2 săptămâni și de 6,1 litere în grupul cu ajustare la 4 săptămâni. Proporția de pacienți la care s-a extins intervalul de tratament la 12 săptămâni sau peste acest interval a fost de 56,9% în grupul cu ajustare la 2 săptămâni și de 60,2% în grupul cu ajustare la 4 săptămâni. La ultima vizită, înainte de săptămâna 96, la 64,9% și 61,2% dintre pacienții din grupul cu ajustare la 2 și respectiv 4 săptămâni a fost planificată următoarea injecție la un interval de 12 săptămâni sau mai mare. În timpul celui de-al doilea an de tratament, pacienților din cele două grupuri cu ajustare la 2 și respectiv 4 săptămâni li s-au administrat în medie 3,6 și, respectiv 3,7 injecții. Pentru perioada de 2 ani de tratament, pacienților li s-a administrat o medie de 10,4 injecții.
Profilurile de siguranță oculară și sistemică au fost similare cu siguranța observată în studiile pivot VIEW1 și VIEW2.
ARIES a fost un studiu cu o durată de 104 săptămâni, multicentric, randomizat, în regim deschis, controlat cu comparator activ efectuat la 269 pacienți naivi la tratament pentru DMLV forma umedă, proiectat pentru a evalua non-inferioritatea în termeni de eficacitate precum și siguranță a unui regim de doze de tipul tratament și extindere inițiat după 3 doze lunare consecutive urmate de o extindere la un interval de tratament de 2 luni versus regimul de doze tip tratament și extindere inițiat după primul an de tratament.
Studiul ARIES a explorat de asemenea și procentul de pacienți care au necesitat tratament mai frecvent de 8 săptămâni pe baza deciziei investigatorului. Din 269 pacienți, 62 pacienți au primit doze mai frecvente cel puțin o dată pe parcursul studiului. Acești pacienți au rămas în studiu și au primit tratament în acord cu evaluarea clinică cea mai bună a investigatorului, dar nu mai frecvent de 4 săptămâni și intervalele de tratament au putut fi extinse din nou ulterior. Intervalul mediu de tratament după decizia de a trata mai frecvent a fost de 6,1 săptămâni. În săptămâna 104 AVOC a fost mai mică la pacienții care au avut nevoie de tratament intensiv cel puțin o dată pe parcursul studiului, comparativ cu pacienții care nu au au avut nevoie, iar schimbarea medie în AVOC la finalul studiului față de nivelul de bază a fost de +2,3 ± 15,6 litere. Printre pacienții tratați mai frecvent, 85,5% și-au menținut vederea, de exemplu au pierdut mai puțin de 15 litere și 19,4% au câștigat 15 litere sau mai multe. Profilul de siguranță pentru pacienții tratați mai frecvent de 8 săptămâni a fost comparabil cu datele de siguranță din studiile VIEW 1 și VIEW 2.
Edem macular secundar OVCR
Siguranța și eficacitatea clinică a aflibercept au fost evaluate în două studii randomizate, multicentrice, cu dublă mascare a formei farmaceutice, controlate cu tratament fictiv, la pacienți cu edem macular secundar OVCR (COPERNICUS și GALILEO cu un număr total de 358 pacienți care au fost tratați și evaluați în vederea stabilirii eficacității (217 cu aflibercept). Vârsta pacienților a variat între 22 ani și 89 ani, cu o medie de 64 ani. În studiile efectuate la pacienți cu OVCR, aproximativ 52% (112/217) dintre pacienții randomizați la tratamentul cu aflibercept aveau vârsta de 65 ani sau mai mult, și aproximativ 18% (38/217) aveau vârsta de 75 ani sau mai mult. În ambele studii, pacienții au fost repartizați randomizat, în raport de 3:2, fie în grupul cu aflibercept 2 mg administrat la intervale de 4 săptămâni (2Q4), fie în grupul de control, pentru a li se administra injecții cu tratament fictiv, la intervale de 4 săptămâni, pentru un total de 6 injecții.
După administrarea unei injecții lunar, timp de 6 luni consecutiv, pacienților li s-a administrat tratament numai dacă au întrunit criteriile specificate în prealabil pentru repetarea tratamentului, cu excepția pacienților din grupul de control din cadrul studiului clinic GALILEO, care au continuat administrarea de tratament fictiv (control față de control) până în săptămâna 52. Începând din acest moment, tuturor pacienților li s-a administrat tratament dacă au întrunit criteriile specificate în prealabil.
În ambele studii, obiectivul principal al eficacității a fost procentul pacienților care a câștigat cel puțin 15 litere din AVOC în săptămâna 24, comparativ cu momentul inițial. A doua variabilă de eficacitate secundară a fost modificarea acuității vizuale în săptămâna 24 comparativ cu momentul inițial.
Diferența între grupurile de tratament a fost favorabilă pentru aflibercept într-un mod semnificativ statistic, în ambele studii. Îmbunătățirea maximă a acuității vizuale s-a realizat la 3 luni cu stabilizarea ulterioară asupra acuității vizuale și asupra GCR până la 6 luni. Diferența semnificativă statistic a fost menținută până în săptămâna 52.
Rezultatele detaliate din analiza ambelor studii sunt prezentate în Tabelul 3 și Figura 2 de mai jos.
Tabelul 3: Rezultatele privind eficacitatea în săptămâna 24, săptămâna 52 și săptămâna 76/100 (Set complet de analiză cu LOCFC)) în studiile COPERNICUS și GALILEO
| Rezultate privind eficacitatea | Studiul COPERNICUS | Studiul GALILEO | ||||||||||
| 24 săptămâni | 52 săptămâni | 100 săptămâni | 24 săptămâni | 52 săptămâni | 76 săptămâni | |||||||
| Aflibercept 2 mg Q4 (N = 114) | Control (N= 73) | Aflibercept 2 mg (N = 114) | Control E) (N =73) | Aflibercept F) 2 mg (N = 114) | Control E,F) (N=73) | Aflibercept 2 mg Q4 (N = 103) | Control (N = 68) | Aflibercepti 2 mg (N = 103) | Control (N = 68) | Aflibercept G) 2 mg (N = 103) | Control G) (N = 68) | |
| Procentul pacienților care au câștigat ≥15 litere din AVOCC) față de momentul inițial | 56% | 12% | 55% | 30% | 49,1% | 23.3% | 60% | 22% | 60% | 32% | 57,3% | 29.4% |
| Diferența ponderatăA,B,E) (IÎ 95%) valoare-p | 44,8% (33,0; 6,6) p < 0,0001 | 25,9% (11,8; 40,1) p = 0,0006 | 26,7% (13,1; 40,3) p=0,0003 | 38,3% (24,4; 52,1) p < 0,0001 | 27,9% (13,0; 42,7) p = 0,0004 | 28,0% (13,3; 42,6) p=0,0004 | ||||||
Modificarea medie în AVOCC) măsurată prin scorul literelor ETDRSC) față de momentul inițial (DS) | 17,3 (12,8) | -4.0 (18.0) | 16,2 (17,4) | 3.8 (17.1) | 13,0 (17,7) | 1.5 (17.7) | 18,0 (12,2) | 3.3 (14.1) | 16,9 (14,8) | 3.8 (18.1) | 13,7 (17,8) | 6.2 (17.7) |
| Diferența în valoarea A,C,D,E) medie a LS (IÎ 95%) valoarea-p | 21,7 (17,4; 6,0) p < 0,0001 | 12,7 (7,7; 17,7) p < 0,0001 | 11,8 (6,7; 17,0) p < 0,0001 | 14.7 (10,8; 18,7) p < 0,0001 | 13,2 (8,2; 18,2) p < 0,0001 | 7,6 (2,1; 13,1) p=0,0070 | ||||||
A. Diferența este reprezentată de tratamentul cu aflibercept 2 mg administrat o dată la 4 săptămâni minus tratament de control
B. Diferența și intervalul de încredere (IÎ) sunt calculate utilizând testul Cochran-Mantel-Haenszel (CMH) ajustat în funcție de regiune (America față de restul lumii pentru studiul clinic COPERNICUS și Europa față de Asia/Pacific pentru studiul clinic GALILEO) și AVOC la momentul inițial (> 20/200 and ≤ 20/200)
C. AVOC: Acuitatea vizuală optim corectată
ETDRS: Early Treatment Diabetic Retinopathy Study (Studiul retinopatiei diabetice cu tratament precoce) LOCF: Extrapolarea în sens longitudinal a ultimelor date observate
DS: Deviație standard
LS: Media celor mai mici pătrate derivată din ANCOVA
17
D. LS diferența în media celor mai mici pătrate și intervalul de încredere (IÎ) pe baza modelului ANCOVA cu factori de tipul: grup de tratament, regiune (America față de restul lumii pentru studiul clinic COPERNICUS și Europa față de Asia/Pacific pentru studiul clinic GALILEO) și AVOC la momentul inițial (> 20/200 și ≤ 20/200)
E. În studiul clinic COPERNICUS, pacienților din grupul de control li s-a putut administra MYNZEPLI la nevoie, la intervale de 4 săptămâni, din săptămâna 24 până în săptămâna 52; pacienții efectuau vizite la interval de 4 săptămâni F. În studiul clinic COPERNICUS, atât pacienților din grupul de control cât și cei cărora li s-a administrat aflibercept 2 mg li s-a putut administra aflibercept 2 mg la nevoie, la intervale de 4 săptămâni, din săptămâna 52 până în săptămâna 96; pacienții efectuau vizite trimestriale obligatorii, dar era posibil să fie consultați cu o frecvență de 4 săptămâni, în cazul în care era necesar. G. În studiul clinic GALILEO, atât pacienților din grupul de control cât și cei cărora li s-a administrat aflibercept 2 mg li s-a putut administra aflibercept 2 mg la nevoie, la intervale de 8 săptămâni, din săptămâna 52 până în săptămâna 68; pacienții efectuau vizite la interval de 8 săptămâni.
18
Figura 2: Modificarea medie de la momentul inițial în săptămâna 76/100 în ceea ce privește acuitatea vizuală în funcție de grupul de tratament pentru studiile COPERNICUS și GALILEO (Set complet de analiză)
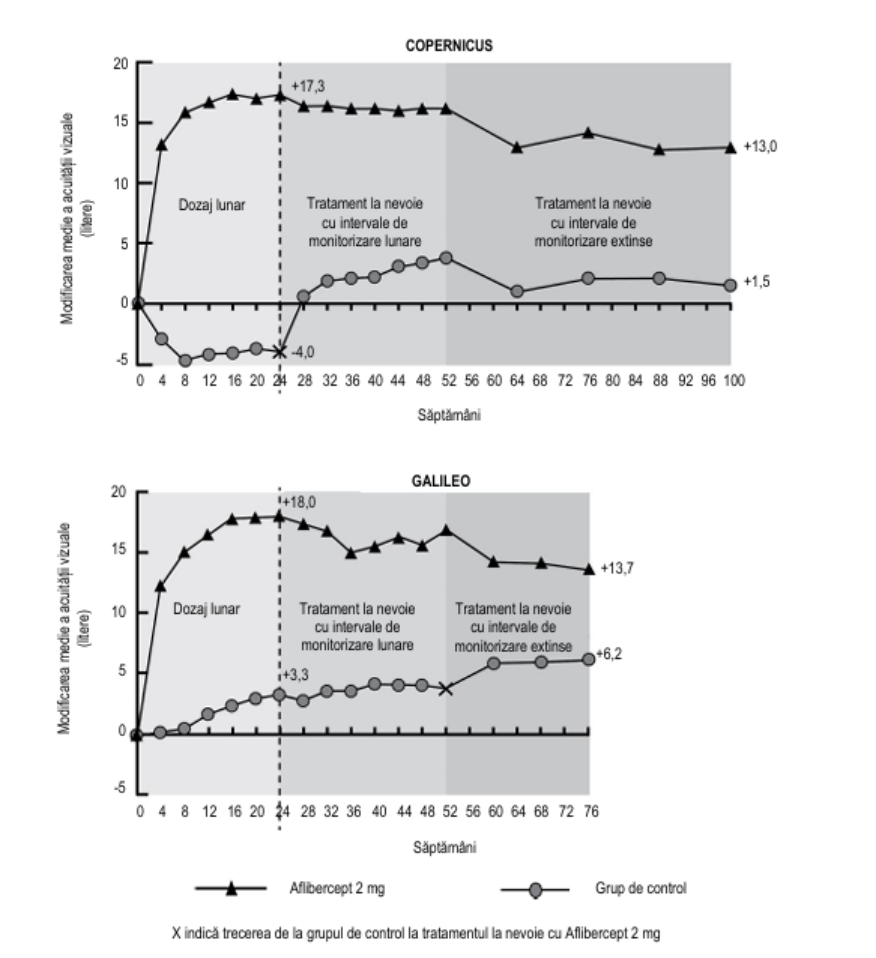
În studiul clinic GALILEO, un procent de 86,4% (n=89) din grupul care a primit tratament cu aflibercept și 79,4% (n = 54) din grupul care a primit tratament fictiv au fost injectați pentru OVCR la momentul inițial. În săptămâna 24, procentul a fost de 91,8% (n = 89) în grupul care a primit tratament cu aflibercept și 85,5% (n = 47) în grupul care a primit tratament fictiv. Aceste proporții s-au menținut în săptămâna 76, cu 84,3% (n = 75) în grupul care a primit tratament cu aflibercept și 84,0% (n = 42) în grupul care a primit tratament fictiv.
În studiul clinic COPERNICUS, un procent de 67,5% (n=77) din grupul care a primit tratament cu aflibercept și 68,5% (n = 50) din grupul care a primit tratament fictiv au fost injectați pentru OVCR la momentul inițial. În săptămâna 24, procentul a fost de 87,4% (n = 90) în grupul care a primit tratament cu aflibercept și 58,6% (n = 34) în grupul care a primit tratament fictiv. Aceste proporții s-au menținut în săptămâna 100, 76,8 % (n = 76) în grupul care a primit tratament cu aflibercept și 78% (n = 39) în grupul care a primit tratament fictiv. Pacienții din grupul care a primit tratament fictiv au fost eligibili pentru a primi aflibercept începând cu săptămâna 24.
Efectul benefic al tratamentului cu aflibercept asupra funcției vizuale a fost similar la grupurile inițiale de pacienți perfuzați și ne-perfuzați. Rezultatele privind eficacitatea în alte subgrupuri, care au putut fi evaluate în fiecare studiu (de exemplu: vârstă, sex, rasă, acuitate vizuală la momentul inițial, durata OVCR), au confirmat rezultatele în populația generală.
În analiza datelor centralizate provenite din studiile clinice GALILEO și COPERNICUS, aflibercept a demonstrat modificări semnificative din punct de vedere clinic față de momentul inițial, conform Chestionarului privind funcția vizuală, al Institutului Național pentru Afecțiuni Oculare (NEI VFQ- 25). Importanța acestor modificări a fost similară celor observate în studiile publicate, corespunzând unui câștig de 15 litere din acuitatea vizuală optim corectată (AVOC).
Edem macular secundar ORVR
Siguranța și eficacitatea aflibercept au fost evaluate printr-un studiu randomizat, multicentric, cu dublu orb, controlat, cu comparator activ, desfășurat la pacienți cu edem macular secundar ORVR (ocluziei de ram venos retinian) (Studiul VIBRANT), inclusiv ocluziei hemiretiniene a venei centrale a retinei. În studiul clinic VIBRANT, un număr total de 181 de pacienți au fost tratați și evaluați privind eficacitatea (la 91 dintre pacienți s-a administrat tratament cu aflibercept). Vârsta pacienților a variat între 42 ani și 94 ani, cu o medie de 65 ani. În cadrul studiului la pacienții cu ORVR, aproximativ 58% (53/91) dintre pacienții randomizați la tratamentul cu aflibercept aveau vârsta de 65 ani sau mai mult, iar aproximativ 23% (21/91) aveau vârsta de 75 ani sau mai mult. În cadrul studiului, pacienții au fost randomizați într-un raport de 1:1, fie pentru a li se administra aflibercept 2 mg la interval de 8 săptămâni - după 1 injecție administrată lunar, timp de 6 luni consecutiv, fie pentru a li se efectua fotocoagulare laser la momentul inițial (grupul de control laser). A fost posibil ca pacienților din grupul de control laser să li se efectueze fotocoagulare laser suplimentară (denumită „tratament laser de salvare“) începând cu săptămâna 12, la un interval minim de 12 săptămâni. Pe baza unor criterii prestabilite pacienții din grupul de tratament laser au primit tratament de salvare cu aflibercept 2 mg începând cu săptămâna 24, administrat la interval de 4 săptămâni, timp de 3 luni consecutiv, urmat de injecții intravitreene la interval de 8 săptămâni.
În studiul clinic VIBRANT, criteriul principal de eficacitate a fost reprezentat de proporția de pacienți care au realizat un câștig de cel puțin 15 litere din AVOC în săptămâna 24 în comparație cu momentul inițial și grupul tratat cu aflibercept a fost superioară grupului de control laser.
Un obiectiv secundar de eficacitate a fost modificarea acuității vizuale în săptămâna 24, comparativ cu valoarea inițială, care a fost semnificativ în favoarea aflibercept, din punct de vedere statistic, în studiul clinic VIBRANT. Evoluția ameliorării vizuale a fost rapidă, iar îmbunătățirea maximă a fost atinsă în luna a-3-a, cu menținerea efectului până în luna 12. În grupul tratat cu laser, la 67 de pacienți li s-a administrat tratament de salvare cu aflibercept începând cu săptămâna 24 (grupul comparator activ/grupul tratat cu aflibercept 2 mg) ceea ce a dus la ameliorarea acuității vizuale cu aproximativ 5 litere din săptămâna 24 până în săptămâna 52.
Rezultate detaliate din analiza studiului clinic VIBRANT sunt prezentate în Tabelul 4 și Figura 3 de mai jos.
Tabelul 4: Rezultatele privind eficacitatea în săptămâna 24 și săptămâna 52 (Set complet de analiză cu LOCFC) în studiul clinic VIBRANT
| Rezultate privind eficacitatea | Studiul VIBRANT | |||
| 24 săptămâni | 52 săptămâni | |||
| Aflibercept 2 mg Q4 (N = 91) | Control Activ (laser) (N = 90) | Aflibercept 2 mg Q8 (N = 91) D) | Control Activ (laser)/ Aflibercept 2 mgE) (N = 90) | |
| Procentul pacienților care au câștigat ≥ 15 litere față de momentul inițial (%) | 52,7% | 26,7% | 57,1% | 41,1% |
| Diferența ponderatăA,B) (IÎ 95%) valoare-p | 26,6% (13,0; 40,1) p=0,0003 | 16,2% (2,0; 30,5) p=0,0296 | ||
| Modificarea medie în AVOC măsurată prin scorul literelor ETDRS față de momentul inițial (DS) | 17,0 (11,9) | 6,9 (12,9) | 17,1 (13,1) | 12,2 (11,9) |
| Diferența în valoarea medie a A,C LS (IÎ 95%) valoare-p | 10,5 (7,1; 14,0) p<0,0001 | 5,2 (1,7; 8,7) p=0,0035F) | ||
A) Diferența este reprezentată de aflibercept 2 mg administrat la interval de 4 săptămâni minus tratament de control activ (laser)
B) Diferența și intervalul de încredere (IÎ) sunt calculate utilizând schema de ponderare Mantel-Haenszel ajustat în funcție de regiune (America față de Japonia) și AVOC la momentul inițial (> 20/200 și ≤ 20/200) C) LS diferența în media celor mai mici pătrate și intervalul de încredere (IÎ 95%) pe baza modelului ANCOVA cu factori de tipul: grup de tratament, AVOC la momentul inițial (> 20/200 și ≤ 20/200) și regiune (America față de Japonia) ca efecte fixe, și AVOC la momentul inițial ca și co-variantă.
D) Din săptămâna 24, intervalul de tratament, al grupului tratat cu aflibercept, a fost extins pentru toți pacienții de la 4 săptămâni la 8 săptămâni în timpul săptămânii 48.
E) Începând cu săptămâna 24, pacienților din grupul tratat cu laser li s-ar putea administra tratament de salvare cu aflibercept, dacă s-a atins cel puțin un criteriu prespecificat de eligibilitate. La un număr total de 67 subiecți din acest grup li s-a administrat tratament cu aflibercept de salvare. Regimul stabilit pentru tratamentul de salvare cu aflibercept a fost de 2 mg la interval de 4 săptămâni, urmate de administrarea unei injecții la interval de 8 săptămâni.
F) Valoare-p nominală
Figura 3: Studiul clinic VIBRANT - Modificarea medie a AVOC măsurată prin scorul literelor ETDRS față
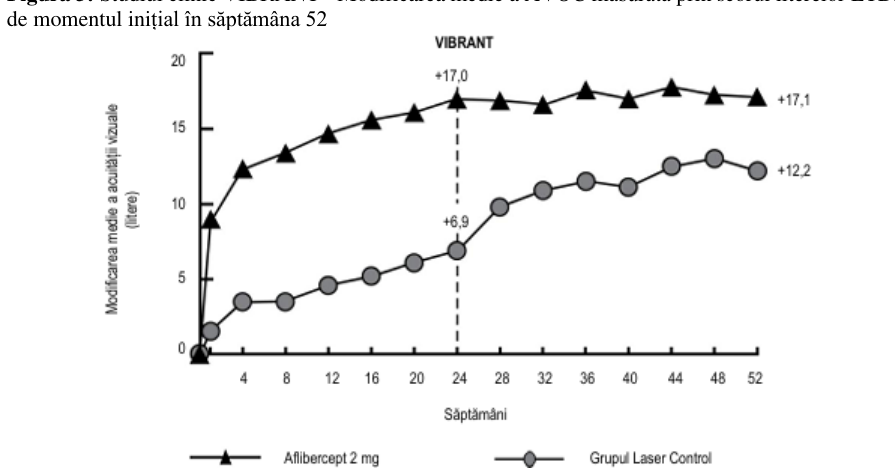
La momentul inițial, proporția pacienților perfuzați în grupul tratat cu aflibercept și grupul tratat cu laser a fost de 60% și, respectiv, 68%. În săptămâna 24, proporția pacienților perfuzați în grupul tratat cu aflibercept și grupul tratat cu laser a fost de 80% și, respectiv, 67%. În grupul tratat cu aflibercept, proporția pacienților perfuzați a fost menținută până la săptămâna 52. În grupul tratat cu laser, în care pacienții au fost eligibili pentru tratamentul cu aflibercept de salvare din săptămâna 24, proporția pacienților perfuzați a crescut la 78% până în săptămâna 52.
Edem macular diabetic
Siguranța și eficacitatea clinică a aflibercept au fost evaluate în două studii clinice randomizate, multicentrice, dublu-orb, controlate activ, la pacienți cu EMD (VIVIDDME și VISTADME). Un total de 862 pacienți au fost tratați și evaluați din punct de vedere al eficacității, 576 cu aflibercept.
Vârsta pacienților a variat între 23 ani și 87 ani, cu o medie de 63 ani. În studiile clinice efectuate cu pacienți cu EMD, aproximativ 47% (268/576) dintre pacienții randomizați la tratamentul cu aflibercept aveau vârsta de 65 ani sau mai mult și aproximativ 9% (52/576) aveau vârsta de 75 ani sau mai mult. Majoritatea pacienților din ambele studii clinice aveau diabet zaharat de tip II.
În ambele studii clinice, pacienții au fost repartizați randomizat, în raport de 1:1:1, pentru unul din cele 3 regimuri de dozare:
- Aflibercept administrat în doză de 2 mg la intervale de 8 săptămâni, după administrarea unei injecții lunare, timp de 5 luni consecutiv (aflibercept 2Q8);
- Aflibercept administrat în doză de 2 mg la intervale de 4 săptămâni (aflibercept 2Q4);
- Foto-coagulare cu laser la nivel macular (control cu tratament activ).
Începând cu săptămâna 24, pacienții care îndeplineau condiția pentru un prag prespecificat de pierdere a vederii erau eligibili pentru a li se administra tratament suplimentar: pacienții din grupurile tratate cu aflibercept puteau fi tratați cu laser, iar pacienții din grupul de control puteau fi tratați cu aflibercept.
În ambele studii clinice, obiectivul principal al eficacității a fost modificarea medie a AVOC de la momentul inițial până în săptămâna 52, atât grupul tratat cu aflibercept 2Q8, cât și grupul tratat cu aflibercept 2Q4 au demonstrat eficacitate superioară statistic față de grupul de control. Acest beneficiu s-a menținut până în săptămâna 100.
Rezultatele detaliate provenind din analiza studiilor clinice VIVIDDME și VISTADME sunt prezentate în Tabelul 5 și Figura 4 de mai jos.
Tabelul 5: Rezultatele privind eficacitatea în săptămâna 52 și săptămâna 100 (Set complet de analiză cu LOCF) în studiile clinice VIVIDDME și
VISTADME
| Rezultate privind eficacitatea | Studiul VIVIDDME | Studiul VISTADME | ||||||||||
| 52 săptămâni | 100 săptămâni | 52 săptămâni | 100 săptămâni | |||||||||
| Aflibercept 2 mg Q8 A (N = 135) | Aflibercept2 mg Q4 (N = 136) | Control activ (laser) (N = 132) | Aflibercep t 2 mg Q8 A (N = 135) | afliberce pt 2 mg Q4 (N = 136) | Control activ (laser) (N = 132) | Aflibercept 2 mg Q8 A (N = 151) | Aflibercept 2 mg Q4 (N = 154) | Control activ (laser) (N = 15 4) | Aflibercept 2 mg Q8 A (N=151) | aflibercep t 2 mg Q4 (N=154) | Control activ (laser) (N=154) | |
| Modificarea medie a AVOC măsurată prin scorul literelor ETDRS E față de momentul inițial | 10,7 | 10,5 | 1,2 | 9,4 | 11,4 | 0,7 | 10,7 | 12,5 | 0,2 | 11,1 | 11,5 | 0,9 |
| Diferența valorii medii a LSB,C,E (IÎ 97,5%) | 9,1 (6,3; 11,8) | 9,3 (6,5; 12,0) | 8,2 (5,2; 11,3) | 10,7 (7,6; 13,8) | 10,45 (7,7; 13,2) | 12,19 (9,4; 15,0) | 10,1 (7,0; 13,3) | 10,6 (7,1; 14,2) | ||||
| Procentul pacienților care au câștigat ≥ 15 litere dinmomentul inițial | 33% | 32% | 9% | 31,1% | 38,2% | 12,1% | 31% | 42% | 8% | 33,1% | 38,3% | 13,0% |
| Diferența ajustată D,C,E (IÎ 97,5%) | 24% (13,5; 34,9) | 23% (12,6; 33,9) | 19,0% (8,0; 29,9) | 26,1% (14,8; 37,5) | 23% (13,5; 33,1) | 34% (24,1; 44,4) | 20,1% (9,6; 30,6) | 25,8% (15,1; 36,6) | ||||
A După inițierea tratamentului cu 1 injecție administrată lunar, timp de 5 luni consecutiv
B Media LS și IÎ pe baza modelului ANCOVA cu determinarea AVOC la momentul inițial drept covariabilă și un factor pentru grupul de tratament. În plus, regiunea (Europa/Australia față de Japonia) a fost inclusă ca factor pentru studiul clinic VIVIDDME, iar antecedentele de IM și/sau AVC au fost incluse ca factor pentru studiul clinic VISTADME.
C Diferența este reprezentată de grupul tratat cu aflibercept minus grupul cu tratament de control activ (laser) D Diferența cu interval de încredere (IÎ) și testul statistic sunt calculate utilizând schema de ponderare Mantel-Haenszel ajustată în funcție de regiune (Europa/Australia față de Japonia) pentru studiul clinic VIVIDDME și antecedentele medicale de IM sau AVC pentru studiul clinic VISTADME E AVOC: Acuitatea vizuală optim corectată
ETDRS: Early Treatment Diabetic Retinopathy Study (Studiul retinopatiei diabetice cu tratament precoce) LOCF: Extrapolarea în sens longitudinal a ultimelor date observate
LS: Media celor mai mici pătrate derivată din ANCOVA IÎ: Interval de încredere
23
Figura 4: Modificarea medie a AVOC măsurată prin scorul literelor ETDRS de la momentul inițial până în săptămâna 100 în studiile clinice VIVIDDME și VISTADME
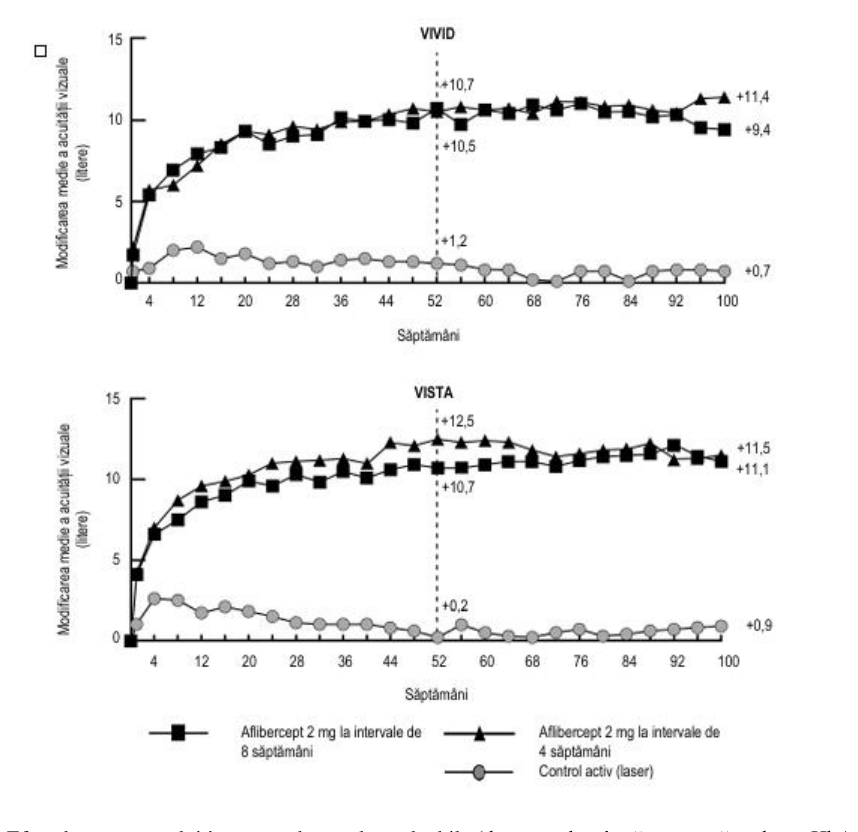
Efectele tratamentului în toate subgrupele evaluabile (de exemplu vârstă, sex, rasă, valoare HbA1c la momentul inițial, acuitate vizuală la momentul inițial, tratament anti-VEGF anterior) în fiecare studiu clinic și în analiza combinată au fost, în general, în concordanță cu rezultatele obținute la populațiile globale.
În studiile clinice VIVIDDME și VISTADME, 36 pacienți (9%) și, respectiv, 197 pacienți (43%) au primit anterior tratament anti-VEGF, cu o perioadă de eliminare cu durata de 3 luni sau mai lungă. Efectele tratamentului la subgrupul de pacienți tratați cu un inhibitor de VEGF au fost similare cu efectele observate la pacienții naivi, care nu fuseseră tratați cu un inhibitor de VEGF.
Pacienții cu patologie bilaterală au fost eligibili pentru a li se administra tratament anti-VEGF la celălalt ochi, dacă medicul a considerat că este necesar acest lucru. În studiul clinic VISTADME, la 217 (70,7%) dintre pacienții cu aflibercept li s-au administrat injecții bilaterale cu aflibercept până în săptămâna 100; în studiul clinic VIVIDDME, la 97 (35,8%) dintre pacienții tratați cu aflibercept li s-a administrat un tratament anti- VEGF diferit la celălalt ochi.
Un studiu clinic comparativ independent (DRCR.net Protocol T) a utilizat un regim de dozare flexibil pe baza unor criterii stricte ale TCO și a criteriilor de re-tratament privind acuitatea vizuală. În grupul care a primit tratament cu aflibercept (n = 224), în săptămâna 52, acest regim a condus la o medie de 9,2 doze administrate la acești pacienți, similar cu numărul de doze administrate în grupul tratat cu aflibercept 2Q8 în studiile clinice VIVIDDME și VISTADME, în timp ce eficacitatea totală a grupului care a primit tratament cu aflibercept în studiul clinic Protocol T a fost comparabilă cu cea a grupului care a primit tratament cu aflibercept 2Q8 în studiile clinice VIVIDDME și VISTADME. În studiul clinic Protocolul T a fost observată o creștere medie de 13,3 litere a acuității vizuale, 42% dintre pacienți câștigând cel puțin 15 litere în acuitatea vizuală față de momentul inițial. Rezultatele privind siguranța au demonstrat că incidența generală a evenimentelor adverse oculare și non-oculare (inclusiv EAT) au fost comparabile în toate grupurile de tratament ale studiilor și între studii.
VIOLET, un studiu clinic de 100 de săptămâni, multicentric, randomizat, în regim deschis, controlat cu comparator activ la pacienți cu EMD a comparat trei regimuri diferite de dozare a aflibercept 2 mg pentru tratamentul EMD după cel puțin un an de tratament la intervale fixe, când tratamentul a fost inițiat cu 5 doze lunare consecutive, urmate de doze la fiecare 2 luni. Studiul a evaluat non- inferioritatea aflibercept 2 mg dozat conform regimului “tratament și extindere” (2T&E când intervalele de injectare au fost ținute la un minimum de 8 săptămâni și extins gradual pe baza rezultatelor clinice și anatomice) și aflibercept 2 mg dozat la nevoie (2PRN când pacienții au fost observați la fiecare 4 săptămâni și le-au fost administrate injecții la nevoie pe baza rezultatelor clinice și anatomice), comparat cu aflibercept 2 mg dozat la fiecare 8 săptămâni (2Q8) pentru al doilea și al treilea an de tratament.
Obiectivul primar de evaluare a eficacității (modificarea AVOC față de nivelul inițial în săptămâna 52) a fost 0,5 ± 6,7 litere în grupul 2T&E și 1,7 ± 6,8 litere în grupul 2PRN comparativ cu 0,4 ± 6,7 litere în grupul 2Q8, obținând non-inferioritate statistică (p<0,0001 pentru ambele comparații; limita NI 4 litere). Modificările AVOC față de nivelul de bază la săptămâna 100 erau consistente cu rezultatele săptămânii 52: - 0,1 ± 9,1 litere în grupul 2T&E și 1.8 ± 9.0 în grupul 2PRN comparat cu 0.1 ± 7.2 litere în grupul 2Q8. Media numărului de injectări peste 100 săptămâni a fost de 12,3, 10,0 și 11,5 pentru 2Q8fix, 2T&E și respectiv 2PRN.
Profilurile de siguranță oculară și sistemică în toate cele 3 grupuri de tratament au fost similare cu cele observate în studiile pivot VIVID și VISTA.
În grupul 2T&E, incrementele și decrementele pentru intervalele de injectare au fost la discreția investigatorului; ajustările de 2 săptămâni au fost recomandate în cadrul studiului.
Neovascularizație coroidală miopică
Siguranța și eficacitatea aflibercept au fost evaluate în cadrul unui studiu randomizat, multicentric, cu dublă mascare a formei farmaceutice, controlat cu tratament fictiv, efectuat la pacienți asiatici cu NVC miopică, netratați anterior. Un total de 121 de pacienți au fost tratați și au fost evaluați din punct de vedere al eficacității (90 cu aflibercept). Vârsta pacienților a variat între 27 ani și 83 ani, cu o medie de 58 ani. În cadrul studiului efectuat la pacienții cu NVC miopică, aproximativ 36% (33/91) dintre pacienții randomizați la tratamentul cu aflibercept aveau vârsta de 65 ani sau mai mult, și aproximativ 10% (9/91) aveau vârsta de 75 ani sau mai mult.
Pacienții au fost repartizați aleator într-un raport de 3:1 pentru a primi fie 2 mg aflibercept intravitrean, fie injecții cu tratament fictiv, administrate la începutul studiului, injecții suplimentare administrându-se lunar în cazul persistenței sau recurenței bolii, până la săptămâna 24, când s-a evaluat obiectivul primar. La săptămâna 24, pacienții randomizați inițial pentru tratamentul fictiv au fost eligibili pentru a primi prima doză de aflibercept. În urma acesteia, pacienții din ambele grupuri au fost eligibili în continuare pentru injecții suplimentare în cazul persistenței sau recurenței bolii.
Diferența dintre grupurile de tratament a fost semnificativă statistic în favoarea aflibercept pentru obiectivul primar (modificarea AVOC) și obiectivul secundar de confirmare privind eficacitatea (proporția de pacienți la care s-a înregistrat un câștig de 15 litere din AVOC) la săptămâna 24 comparativ cu momentul inițial. Diferențele pentru ambele obiective s-au menținut până la săptămâna 48.
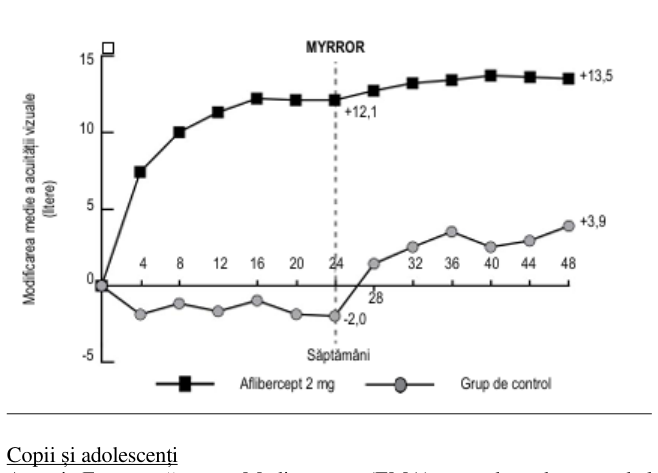
Rezultatele detaliate ale analizei din cadrul studiului clinic MYRROR sunt prezentate în Tabelul 6 și Figura 5 de mai jos.
Tabelul 6: Rezultatele privind eficacitatea în săptămâna 24 (analiza primară) și săptămâna 48 în studiul clinic MYRROR (Set complet de analiză cu LOCFA))
| Rezultate privind eficacitatea | Studiul MYRROR | |||
| 24 săptămâni | 48 săptămâni | |||
| Aflibercept 2 mg (N = 90) | Tratament fictiv (N = 31) | Aflibercept 2 mg (N = 90) | Tratament fictiv/ Aflibercept 2 mg (N = 31) | |
| Modificarea medie a AVOCB)măsurată prin scorul literelor ETDRS față de momentul inițial (DS) E) | 12,1 (8,3) | -2,0 (9,7) | 13,5 (8,8) | 3,9 (14,3) |
| Diferența în valoarea medie a LSC,D,E) (IÎ 95%) | 14,1 (10,8; 17,4) | 9,5 (5,4; 13,7) | ||
| Proporția de pacienți cu un câștig ≥15 litere de la momentul inițial | 38,9% | 9,7% | 50,0% | 29,0% |
| Diferența ponderată D,F) (IÎ 95%) | 29,2% (14,4; 44,0) | 21,0% (1,9; 40,1) | ||
A) LOCF: Extrapolarea în sens longitudinal a ultimelor date observate
B) AVOC Acuitatea vizuală optim corectată
ETDRS: Early Treatment Diabetic Retinopathy Study (Studiul retinopatiei diabetice cu tratament precoce) DS: Deviație standard
C) Media LS: Media celor mai mici pătrate derivată din ANCOVA
D) IÎ: Interval de încredere
E) Diferența în valoarea medie LS și IÎ 95% pe baza unui model ANCOVA cu factori de tipul grup de tratament și țară (indicativele țărilor) și valoarea AVOC de la momentul inițial drept covariabilă. F) Diferența și IÎ 95% CI sunt calculate utilizând testul Cochran-Mantel-Haenszel (CMH) ajustat pentru țară (indicativele țărilor)
Figura 5: Modificarea Medie a Acuității Vizuale în Funcție de Grupul de Tratament, de la Momentul Inițial până în Săptămâna 48, în Studiul Clinic MYRROR (Set complet de analiză, LOCF)
Agenția Europeană pentru Medicamente (EMA) a acordat o derogare de la obligația de depunere a rezultatelor studiilor efectuate cu aflibercept la toate subgrupele de copii și adolescenți în DMLV forma umedă, OVCR, ORVR, EMD și subgrupe în NVC miopică (vezi pct. 4.2 pentru informații privind utilizarea la copii și adolescenți)..
5.2 Proprietăți farmacocinetice
MYNZEPLI se administrează direct în corpul vitros, pentru a exercita efecte locale asupra ochiului.
Absorbție/distribuție
După administrarea intravitreană, aflibercept este absorbit lent de la nivelul ochiului în circulația sistemică și se observă predominant în circulația sistemică sub forma unui complex inactiv, stabil cu VEGF; cu toate acestea, numai „aflibercept liber” se poate lega de VEGF endogen.
Într-un sub-studiu farmacocinetic efectuat la 6 pacienți cu DMLV forma neovasculară (umedă) la care s-au recoltat frecvent probe, concentrațiile plasmatice maxime ale aflibercept liber (Cmax sistemică) au fost foarte scăzute, cu o medie de aproximativ 0,02 micrograme/ml (cuprinsă între 0 și 0,054) în decurs de 1 - 3 zile după injectarea intravitreană a dozei de 2 mg și nu au mai fost detectabile la două săptămâni după administrarea dozei, la aproape toți pacienții. Aflibercept nu se acumulează în plasmă când este administrat intravitrean la interval de 4 săptămâni.
Concentrația plasmatică maximă a aflibercept liber este de aproximativ 50 - 500 de ori mai mică decât concentrația de aflibercept necesară pentru inhibarea activității biologice a VEGF sistemic cu 50%, la modele animale la care s-au observat modificări ale tensiunii arteriale, după ce valorile circulante ale aflibercept liber au fost de aproximativ 10 micrograme/ml și au revenit la valorile inițiale, după ce valorile circulante ale aflibercept liber au scăzut subaproximativ 1 microgram/ml. Se estimează că după administrarea intravitreană a dozei de 2 mg la pacienți, concentrația plasmatică maximă medie a aflibercept liber este de peste 100 de ori mai mică decât concentrația aflibercept necesară pentru legarea maximă a 50% din VEGF sistemic (2,91 micrograme/ml) într- un studiu efectuat cu voluntari sănătoși. Prin urmare, efectele farmacodinamice sistemice, cum sunt modificările tensiunii arteriale, sunt improbabile.
În cadrul unor sub-studii farmacocinetice la pacienți cu OVCR, ORVR, EMD sau NVC miopică valoarea medie a Cmax de aflibercept liber în plasmă a fost similară cu valorile din intervalul de 0,03 – 0,05 micrograme/ml și intervalul individual nu a depășit 0,14 micrograme/ml. După aceea, concentrațiile plasmatice pentru aflibercept liber au scăzut de la valori mai mici sau aproape de limita inferioară de cuantificare, în general, într-o săptămână; după 4 săptămâni, înainte de administrarea următoare, au fost atinse concentrații nedetectabile la toți pacienții.
Eliminare
Având în vedere faptul că MYNZEPLI este un medicament pe bază de proteine, nu s-au efectuat studii privind metabolizarea.
Aflibercept liber se leagă de VEGF pentru a forma un complex inert, stabil. Similar altor proteine cu molecule mari, se anticipează că atât aflibercept liber cât și cel legat vor fi eliminate prin catabolism proteolitic.
Insuficiență renală
Nu s-au efectuat studii specifice cu aflibercept la pacienți cu insuficiență renală.
Analiza farmacocinetică la pacienții incluși în studiul clinic VIEW2, dintre care 40% aveau insuficiență renală (24% ușoară, 15% moderată și 1% severă), nu a sugerat nicio diferență în ceea ce privește concentrațiile plasmatice ale medicamentului activ după administrarea intravitreană la intervale de 4 sau 8 săptămâni.
Rezultate similare au fost observate la pacienții cu OVCR în cadrul studiului clinic GALILEO, la pacienții cu EMD în cadrul studiului clinic VIVIDDME și la pacienții cu NVC miopică în studiul clinic MYRROR.
5.3 Date preclinice de siguranță
În studiile pre-clinice, au fost observate efecte de toxicitate la doze repetate numai la expuneri sistemice considerate substanțial mai mari față de expunerea maximă la om după administrarea intravitreană în dozele clinice stabilite, fapt ce indică o relevanță clinică scăzută.
La maimuțe, cărora li s-a administrat aflibercept intravitrean, au fost observate eroziuni și ulcerații ale epiteliului respirator al cornetelor nazale la expuneri sistemice mai mari față de expunerea maximă la om. La o valoare a concentrației la care nu se observă nicio reacție adversă (NOAEL – No Observed Adverse effect Level), de 0,5 mg/ochi la maimuțe, expunerea sistemică pentru aflibercept liber a fost de 42 și de 56 de ori mai mare, pe baza Cmax și ASC, comparativ cu valorile corespunzătoare observate la pacienți adulți.
Nu s-au efectuat studii privind potențialul mutagen sau carcinogen al aflibercept.
În cadrul studiilor privind dezvoltarea embriofetală la femele gestante de iepure, s-a demonstrat un efect al aflibercept asupra dezvoltării intrauterine în cazul administrării intravenoase (3 – 60 mg/kg) și subcutanate (0,1 mg/kg – 1 mg/kg). Valoarea la care nu se observă reacții adverse (NOAEL) materne a apărut la o doză de 3 mg/kg și, respectiv, de 1 mg/kg. Nu a fost identificat NOAEL legat de dezvoltare. La doza de 0,1 mg/kg, expunerea sistemică pe baza Cmax și ASC cumulativă pentru aflibercept liber a fost de aproximativ 17 și, respectiv, de 10 ori mai mare, comparativ cu valorile corespunzătoare observate la om după administrarea intravitreană a unei doze de 2 mg.
Efectele asupra fertilității masculine și feminine au fost evaluate în cadrul unui studiu cu durata de 6 luni, efectuat la maimuțe la care s-a administrat intravenos aflibercept în doze cuprinse între 3 și 30 mg/kg. La toate dozele s-au observat menstruații absente sau neregulate asociate cu modificări ale concentrațiilor hormonilor sexuali la femele și modificări ale morfologiei și motilității spermatozoizilor. Pe baza Cmax și ASC pentru aflibercept liber observate la administrarea de doze intravenoase de 3 mg/kg, expunerile sistemice au fost de aproximativ 4900 și, respectiv, de 1500 de ori mai mari comparativ cu valorile corespunzătoare observate la om după administrarea intravitreană a unei doze de 2 mg. Toate modificările au fost reversibile.
6. PROPRIETĂȚI FARMACEUTICE
6.1 Lista excipienților
L-histidină
Clorhidrat de L-histidină monohidrat
Trehaloză dihidrat
Poloxamer 188
Apă pentru preparate injectabile
6.2 Incompatibilități
În absența studiilor de compatibilitate, acest medicament nu trebuie amestecat cu alte medicamente.
6.3 Perioada de valabilitate
2 ani
6.4 Precauții speciale pentru păstrare
A se păstra la frigider (2°C - 8°C). A nu se congela.
A se păstra în ambalajul original pentru a fi protejat de lumină.
Blisterul nedeschis poate fi păstrat în afara frigiderului, sub 25°C timp de cel mult 24 ore. Utilizând o tehnică aseptică, se scoate seringa din blisterul sterilizat.
6.5 Natura și conținutul ambalajului
Soluție în seringă preumplută (sticlă de tip I) marcată cu o linie de dozare, prevăzută cu un dop cu piston (din cauciuc elastomeric brombutilic) și un adaptor de tip Luer Lock cu capac fără filet (din cauciuc elastomeric). Fiecare seringă preumplută conține un volum extractibil de cel puțin 0,09 ml. Mărimea ambalajului este de 1 seringă preumplută.
6.6 Precauții speciale pentru eliminarea reziduurilor și alte instrucțiuni de manipulare
Seringa preumplută este pentru utilizare unică, într-un singur ochi. Extragerea mai multor doze dintr-o seringă preumplută poate crește riscul de contaminare și infecție ulterioară.
A nu se deschide blisterul conținând seringa preumplută sterilă decât într-o încăpere curată, special destinată administrării. Orice medicament neutilizat sau material rezidual trebuie eliminat în conformitate cu reglementările locale.
Seringa preumplută conține mai mult decât doza recomandată de aflibercept 2 mg (echivalent cu 0,05 ml) pentru pacienți. Vezi punctul următor „Instrucțiuni pentru utilizarea seringii preumplute”.
Înainte de administrare soluția trebuie inspectată vizual pentru a observa particulele străine și/sau modificări de culoare sau orice variație a aspectului fizic. În cazul în care se observă particule sau dacă soluția este tulbure sau prezintă modificări de culoare, medicamentul se aruncă.
Pentru injectarea intravitreană trebuie utilizat un ac pentru injectare de 30 G x ½ inch.
Instrucțiuni pentru utilizarea seringii preumplute:
Pentru a pregăti seringa preumplută pentru administrare, urmați toți pașii de mai jos.
1 Când este pregătită administrarea MYNZEPLI, se deschide cutia și se scoate blisterul sterilizat. Se scoate cu atenție pelicula blisterului, asigurând-se menținerea sterilității conținutului acestuia. Se păstrează seringa în plăcuța sterilă până când este pregătită pentru asamblare.
2 Utilizând o tehnică aseptică, se scoate seringa din blisterul sterilizat.
3
4 Pentru a evita compromiterea sterilității medicamentului, nu se împinge pistonul înapoi.
5
6 Ținând seringa cu acul orientat în sus, se verifică prezența bulelelor de aer în seringă. Dacă există bule de aer, se bate ușor seringa cu vârful degetelor până când bulele de aer se ridică la suprafață.
7 Volumul în exces trebuie eliminat înainte de administrare. Eliminați toate bulele și excesul de medicament, apăsând încet pistonul pentru a alinia baza boltei pistonului (nu vârful boltei pistonului) cu linia de marcare a dozei de pe seringă (echivalentă cu 0,05 ml, adică aflibercept 2 mg).
Notă: Această poziționare precisă a pistonului este foarte importantă, deoarece poziționarea incorectă a pistonului poate duce la eliberarea unei cantități mai mari sau mai mici decât doza recomandată
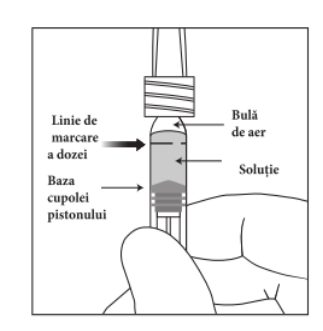
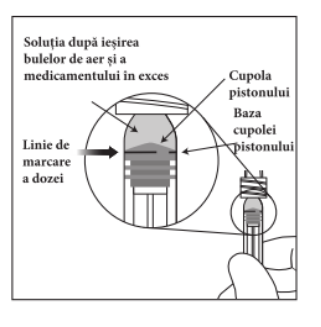
8 Injectați în timp ce apăsați pistonul cu atenție și presiune constantă. Nu aplicați presiune suplimentară odată ce pistonul a ajuns la capătul seringii. Nu administrați nicio cantitate de soluție reziduală observată în seringă.
9 Seringa preumplută este numai de unică folosință. Extragerea dozelor multiple din seringa preumplută poate crește riscul de contaminare și ulterior, infecție.
Orice medicament neutilizat sau material rezidual trebuie eliminat în conformitate cu reglementările locale.
7. DEȚINĂTORUL AUTORIZAȚIEI DE PUNERE PE PIAȚĂ
Advanz Pharma Limited
Unit 17 Northwood House
Northwood Crescent
Dublin 9
D09 V504
Irlanda
8. NUMĂRUL(ELE) AUTORIZAȚIEI DE PUNERE PE PIAȚĂ
EU/1/25/1964/002
9. DATA PRIMEI AUTORIZĂRI SAU A REÎNNOIRII AUTORIZAȚIEI
Data primei autorizări: 18 august 2025
10. DATA REVIZUIRII TEXTULUI
Informații detaliate privind acest medicament sunt disponibile pe site-ul Agenției Europene pentru Medicamente (EMA) https://www.ema.europa.eu.
Acest medicament face obiectul unei monitorizări suplimentare. Acest lucru va permite identificarea rapidă de noi informații referitoare la siguranță. Profesioniștii din domeniul sănătății sunt rugați să raporteze orice reacții adverse suspectate. Vezi pct. 4.8 pentru modul de raportare a reacțiilor adverse
1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI
MYNZEPLI 40 mg/ml soluție injectabilă în flacon
2. COMPOZIȚIA CALITATIVĂ ȘI CANTITATIVĂ
1 ml de soluție injectabilă conține aflibercept* 40 mg.
Un flacon conține un volum ce poate fi extras de cel puțin 0,1 ml, echivalent cu aflibercept cel puțin 4 mg. Acesta furnizează o cantitate utilizabilă pentru administrarea unei doze unice de 0,05 ml, conținând aflibercept 2 mg.
*Proteina de fuziune este formată din fragmente VEGF uman (factor endotelial de creștere vasculară) din domeniile extracelulare ale receptorilor 1 și 2 fuzionate cu fragmentul Fc a IgG1 uman obținută, prin tehnologie ADN recombinantă, în celule ovariene de hamster chinezesc (CHO) K1.
Pentru lista tuturor excipienților, vezi pct. 6.1.
3. FORMA FARMACEUTICĂ
Soluție injectabilă (injecție)
Soluție sterilă, limpede, incoloră până la galben pal, izoosmotică.
4. DATE CLINICE
4.1 Indicații terapeutice
MYNZEPLI este indicat la adulți pentru tratamentul:
- degenerescenței maculare legată de vârstă (DMLV) forma neovasculară (umedă) (vezi pct. 5.1),
- afectării acuității vizuale determinată de edemul macular secundar ocluziei venei retinei (OVR de ram sau OVR centrală) (vezi pct. 5.1),
- afectării acuității vizuale determinată de edemul macular diabetic (EMD) (vezi pct. 5.1),
- afectării acuității vizuale determinată de neovascularizația coroidală miopică (NVC miopică) (vezi pct. 5.1).
4.2 Doze și mod de administrare
MYNZEPLI se administrează numai sub formă de injecții intravitreene.
MYNZEPLI trebuie administrat numai de către un medic oftalmolog cu experiență în administrarea injecțiilor intravitreene.
Doze
DMLV forma umedă
Doza recomandată de MYNZEPLI este de 2 mg aflibercept, echivalent cu 0,05 ml.
Tratamentul cu MYNZEPLI este inițiat cu o injecție o dată pe lună pentru trei administrări consecutive. Intervalul de tratament este apoi extins la două luni.
Pe baza interpretării de către medic asupra rezultatelor funcției vizuale și/sau modificărilor anatomice, intervalul de tratament poate fi menținut la două luni sau extins suplimentar, cu un regim de dozaj de tip „tratament și extindere”, crescând intervalele de injectare în incrementuri de 2 sau 4 săptămâni, astfel încât rezultatele vizuale și/sau anatomice să fie menținute stabile.
În cazul în care rezultatele vizuale și/sau anatomice se deteriorează, intervalul de administrare a tratamentului trebuie scăzut în mod corespunzător.
Nu există nicio cerință pentru monitorizarea între injectări. În funcție de evaluarea medicului, programul vizitelor de monitorizare poate avea o frecvență mai mare decât cel al vizitelor pentru injectare.
Nu au fost studiate intervale de tratament între injectări mai mari de patru luni sau mai mici de patru săptămâni (vezi pct. 5.1).
Edem macular secundar OVR (OVR de ram sau OVR centrală)
Doza recomandată de MYNZEPLI este de 2 mg aflibercept, echivalent cu 0,05 ml.
După injectarea inițială, tratamentul este administrat lunar. Intervalul dintre 2 doze nu trebuie să fie mai mic de o lună.
În cazul în care rezultatele vizuale și anatomice indică faptul că pacientul nu prezintă beneficii în cadrul tratamentului continuu, MYNZEPLI trebuie întrerupt.
Tratamentul lunar continuă până când se obține acuitatea vizuală maximă și/sau nu există semne de activitate a bolii. Poate fi necesară administrarea lunară timp de trei luni consecutiv sau mai mult.
Tratamentul poate fi continuat cu un regim de tip „tratament și extindere“, crescând progresiv intervalele de administrare a tratamentului, astfel încât rezultatele vizuale și/sau anatomice să fie menținute stabile, însă nu există date suficiente pentru a concluziona referitor la durata acestui interval. În cazul în care rezultatele vizuale și/sau anatomice se deteriorează, intervalul de administrare a tratamentului trebuie scăzut în mod corespunzător.
Schema de monitorizare și tratament trebuie stabilită de către medicul curant în funcție de răspunsul individual al pacientului.
Monitorizarea activității bolii poate include examen clinic, teste funcționale sau tehnici imagistice (ex. tomografie în coerență optică sau angiofluorografie).
Edem macular diabetic
Doza recomandată de MYNZEPLI este de 2 mg aflibercept, echivalent cu 0,05 ml.
Tratamentul cu MYNZEPLI este început cu o injecție o dată pe lună pentru cinci administrări consecutive, urmat de o injecție la interval de două luni.
Pe baza interpretării de către medic a rezultatelor funcției vizuale și/sau modificărilor anatomice, intervalul de tratament poate fi menținut la 2 luni sau individualizat, cu un regim de tip „tratament și extindere”, crescând de regulă intervalul de administrare a tratamentului cu incrementuri de 2 săptămâni, astfel încât rezultatele vizuale și/sau anatomice să fie menținute stabile. Există date limitate pentru intervale de tratament mai mari de 4 luni În cazul în care rezultatele vizuale și/sau anatomice se deteriorează, intervalul de administrare a tratamentului trebuie scăzut în mod corespunzător. Intervalele de tratament mai scurte de 4 săptămâni nu au fost studiate (vezi pct 5.1). Programul de monitorizare trebuie să fie stabilit de către medicul curant.
Dacă rezultatele vizuale și anatomice indică faptul că pacientul nu prezintă beneficii prin continuarea tratamentului, administrarea MYNZEPLI trebuie oprită.
Neovascularizația coroidală miopică
Doza recomandată de MYNZEPLI este de o singură injecție intravitreană de aflibercept 2 mg echivalent cu 0,05 ml.
Pot fi administrate doze suplimentare dacă rezultatele vizuale și/sau anatomice indică faptul că boala persistă. Recurențele trebuie tratate drept o nouă manifestare a bolii.
Programul de monitorizare trebuie stabilit de către medicul curant. Intervalul dintre două doze nu trebuie să fie mai scurt de o lună.
Grupe speciale de pacienți
Insuficiență hepatică și/sau renală
Nu s-au efectuat studii specifice cu aflibercept la pacienți cu insuficiență hepatică și/sau renală.
Datele disponibile nu sugerează necesitatea ajustării dozei de aflibercept la acești pacienți (vezi pct. 5.2).
Vârstnici
Nu sunt necesare precauții speciale. Există experiență limitată privind utilizarea la pacienți cu vârsta peste 75 ani cu EMD.
Copii și adolescenți
Siguranța și eficacitatea aflibercept nu au fost stabilite la copii și adolescenți. Nu există date relevante pentru utilizarea aflibercept la copii și adolescenți pentru indicațiile DMLV forma umedă, OVCR, ORVR, EMD și NVC miopică.
Mod de administrare
Injecțiile intravitreene trebuie efectuate de către un medic oftalmolog cu experiență în administrarea injecțiilor intravitreene, conform standardelor medicale și ghidurilor în vigoare. În general, trebuie să se asigure condiții adecvate de anestezie și asepsie, inclusiv administrarea locală a unui bactericid cu spectru larg (de exemplu povidonă iodată aplicată la nivelul pielii perioculare, pleoapei și suprafeței oculare). Se recomandă dezinfecția chirurgicală a mâinilor, utilizarea mănușilor sterile, a unor câmpuri sterile și a unui specul de pleoape steril (sau echivalent).
Acul pentru injectare trebuie introdus la 3,5-4,0 mm în spatele limbului, în cavitatea vitroasă, evitându-se meridianul orizontal în direcția centrului globului ocular. Apoi se injectează un volum de 0,05 ml; pentru următoarele injectări trebuie utilizată o altă zonă sclerală.
Imediat după injectarea intravitreană, pacienții trebuie monitorizați pentru creșterea presiunii intraoculare. Monitorizarea adecvată poate consta în verificarea perfuzării nervului optic sau tonometrie. Dacă este necesar, trebuie să fie disponibil echipament steril pentru paracenteza camerei anterioare.
După injectarea intravitreană, pacienții trebuie instruiți să raporteze fără întârziere orice simptome sugestive de endoftalmită (de exemplu durere oculară, înroșirea ochiului, fotofobie, vedere încețoșată).
Fiecare flacon trebuie utilizat numai pentru tratamentul unui singur ochi. Extragerea dozelor multiple dintrun singur flacon poate crește riscul de contaminare și ulterior, infecție.
Flaconul conține mai mult decât doza recomandată de aflibercept 2 mg (echivalent cu 0,05 ml soluție injectabilă). Volumul extractibil dintr-un flacon este volumul care poate fi extras din flacon și nu se foloseṣte în totalitate. Pentru MYNZEPLI în flacon, volumul extractibil este de cel puțin 0,1 ml. Volumul în exces trebuie eliminat înainte de injectarea dozei recomandate (vezi pct. 6.6).
Injectarea întregului volum al flaconului poate duce la supradozaj. Pentru a elimina bulele de aer din flacon împreună cu volumul în exces de medicament se va împinge pistonul astfel încât marginea plată a pistonului să se alinieze cu linia ce marchează 0,05 ml pe seringă (echivalent cu 0,05 ml, adică aflibercept 2 mg) (vezi pct. 4.9 și 6.6).
După injectare, orice medicament neutilizat trebuie eliminat.
Pentru manipularea medicamentului înainte de administrare, vezi pct. 6.6.
4.3 Contraindicații
Hipersensibilitate la substanța activă sau la oricare dintre excipienții enumerați la pct. 6.1.
Infecție oculară sau perioculară activă sau suspectată.
Inflamație intraoculară activă, severă.
4.4 Atenționări și precauții speciale pentru utilizare
Trasabilitate
Pentru a avea sub control trasabilitatea medicamentelor biologice, numele și numărul lotului medicamentului administrat trebuie înregistrate cu atenție.
Reacții asociate injectării intravitreene
Injecțiile intravitreene, inclusiv cele cu aflibercept, au fost asociate cu endoftalmită, inflamație intraoculară, dezlipire regmatogenă de retină, ruptură de retină și cataractă traumatică iatrogenă (vezi pct. 4.8). Atunci când se administrează MYNZEPLI, trebuie utilizate întotdeauna tehnici de injectare aseptice adecvate. În plus, pacienții trebuie monitorizați în timpul săptămânii după injectare, pentru a permite tratamentul precoce în cazul apariției unei infecții. Pacienții trebuie instruiți să raporteze fără întârziere orice simptom care sugerează o endoftalmită sau oricare dintre evenimentele menționate mai sus.
Flaconul conține mai mult decât doza recomandată de aflibercept 2 mg (echivalent cu 0,05 ml). Volumul în exces trebuie eliminat înainte de administrare (vezi pct. 4.2 și 6.6).
S-au observat creșteri ale presiunii intraoculare în decurs de 60 de minute de la administrarea unei injecții intravitreene, inclusiv la cele cu aflibercept (vezi pct. 4.8). Sunt necesare precauții speciale la pacienți cu glaucom insuficient controlat prin tratament (nu se injectează aflibercept dacă presiunea intraoculară este ≥ 30 mmHg). În toate cazurile, atât presiunea intraoculară cât și perfuzia la nivelul rădăcinii nervului optic trebuie monitorizate și tratate corespunzător.
Imunogenitate
Deoarece este o proteină folosită în scop terapeutic există potențial de imunogenitate în cazul administrării afliberceptului (vezi pct. 4.8). Pacienții trebuie instruiți să raporteze orice semn sau simptom de inflamație intraoculară, de exemplu durere, fotofobie sau roṣeață, care ar putea fi semne clinice atribuite hipersensibilității.
Efecte sistemice
Reacțiile adverse sistemice includ hemoragii altele decât cele oculare și evenimente tromboembolice arteriale care au fost raportate după administrarea intravitreană a inhibitorilor VEGF și există un risc teoretic ca acestea să fie legate de inhibarea VEGF. Există date limitate privind siguranța tratamentului la pacienții cu OVCR, ORVR, EMD sau NVC miopică cu antecedente de accident vascular cerebral sau accidente ischemice tranzitorii, sau infarct miocardic în ultimele 6 luni. Trebuie exercitată precauție în cazul în care sunt tratați acești pacienți.
Alte informații:
Similar altor tratamente intravitreene anti-VEGF pentru DMLV forma umedă, OVCR, ORVR, EMD și NVC miopică, următoarele informații sunt de asemenea valabile:
- Siguranța și eficacitatea tratamentului cu aflibercept administrat concomitent la ambii ochi nu au fost studiate în mod sistematic (vezi pct. 5.1). Dacă tratamentul bilateral este efectuat în același timp, acest lucru poate duce la o expunere sistemică crescută ceea ce poate crește riscul reacțiilor adverse sistemice.
- Utilizarea concomitentă a altor anti-VEGF (factorul de creștere al endoteliului vascular)
- Nu există date disponibile referitoare la utilizarea concomitentă a afliberceptului cu alte medicamente anti-VEGF (sistemice sau oculare).
- Factorii de risc asociați cu apariția unei rupturi la nivelul epiteliului pigmentar al retinei după tratamentul anti-VEGF pentru DMLV forma umedă includ detașarea mare și/sau profundă a epiteliului pigmentar al retinei. Inițierea tratamentului cu aflibercept trebuie efectuată cu precauție la pacienții care prezintă acești factori de risc privind rupturile epiteliului pigmentar al retinei.
- Tratamentul trebuie întrerupt la pacienții cu dezlipire regmatogenă de retină sau cu perforații maculare în stadiul 3 sau 4.
- În cazul rupturii de retină tratamentul trebuie întrerupt și nu trebuie reluat până la vindecarea țesutului epitelial al retinei.
- Tratamentul trebuie întrerupt și nu trebuie reluat mai devreme de momentul în care este programată administrarea următoarei doze în cazul:
- scăderii celei mai bune acuități vizuale corectate (BAVC) cu ≥ 30 litere comparativ cu ultima verificare a acuității vizuale,
- unei hemoragii subretiniene care a inclus centrul foveei, sau, dacă suprafața hemoragiei este ≥ 50 % din toată aria lezată.
- Tratamentul trebuie întrerupt și nu trebuie reluat mai devreme de 28 zile înainte sau după planificarea sau efectuarea unei intervenții chirurgicale intraoculare.
- Aflibercept nu trebuie utilizat în timpul sarcinii, cu excepția cazului în care beneficiile potențiale depășesc riscul potențial pentru făt (vezi pct. 4.6).
- Femeile aflate la vârsta fertilă trebuie să utilizeze măsuri contraceptive eficace în timpul tratamentului și timp de cel puțin 3 luni după ultima injecție intravitreană cu aflibercept (vezi pct. 4.6).
- Experiența clinică în tratamentul pacienților cu OVCR și ORVR ischemic este limitată. Tratamentul nu este recomandat la pacienții cu semne clinice de pierdere ireversibilă ischemică a funcției vizuale.
Grupe de pacienți pentru care există date limitate
Există doar o experiență limitată în ceea ce privește tratamentul pacienților cu EMD determinat de diabetul zaharat de tip I sau al pacienților diabetici cu o valoare a HbA1c peste 12 % sau cu retinopatie diabetică proliferativă.
Aflibercept nu a fost studiat la pacienții cu infecții sistemice active sau la pacienții cu afecțiuni concomitente oculare, cum ar fi dezlipirea de retină sau gaura maculară. De asemenea, nu există experiență în ceea ce privește tratamentul cu aflibercept la pacienții diabetici cu hipertensiune arterială necontrolată. Această lipsă a informațiilor trebuie luată în considerare de către medic atunci când tratează acești pacienți.
Pentru NVC miopică, nu există experiență privind utilizarea afliberceptului în tratamentul pacienților care nu aparțin rasei galbene, al pacienților la care s-a efectuat anterior tratament pentru NVC miopică și al pacienților cu leziuni extrafoveale.
Informații referitoare la excipienți
Acest medicament conține sodiu mai puțin de 1 mmol (23 mg) per doză, adică practic „nu conține sodiu”
4.5 Interacțiuni cu alte medicamente și alte forme de interacțiune
Nu s-au efectuat studii privind interacțiunile.
Nu s-a studiat utilizarea suplimentară a tratamentului fotodinamic (TFD) cu verteporfină și aflibercept, deci un profil de siguranță nu este stabilit încă.
4.6 Fertilitatea, sarcina și alăptarea
Femei aflate la vârsta fertilă
Femeile aflate la vârsta fertilă trebuie să utilizeze măsuri contraceptive eficace în timpul tratamentului și timp de cel puțin 3 luni după ultima injecție intravitreană cu aflibercept (vezi pct. 4.4).
Sarcina
Nu există date privind utilizarea aflibercept la femeile gravide. Studiile la animale au evidențiat toxicitate embriofetală (vezi pct. 5.3).
Cu toate că expunerea sistemică după administrarea oculară este foarte scăzută, aflibercept nu trebuie utilizat în timpul sarcinii, cu excepția cazului în care beneficiile potențiale depășesc riscul potențial pentru făt.
Alăptarea
Pe baza unor date foarte limitate la om, aflibercept poate fi excretat în laptele uman la niveluri scăzute. Aflibercept este o moleculă proteică mare și cantitatea de medicament absorbită de copil este de așteptat să fie minimă. Efectele aflibercept asupra nou-născutului/sugarului alăptat sunt necunoscute.
Ca o măsură de precauție, alăptarea nu este recomandată în timpul utilizării MYNZEPLI.
Fertilitatea
Rezultatele provenite din studiile la animale, constând în expunere sistemică crescută indică faptul că aflibercept poate afecta fertilitatea masculină și feminină (vezi pct. 5.3). Nu se anticipează asemenea efecte după administrarea oculară și expunere sistemică foarte scăzută.
4.7 Efecte asupra capacității de a conduce vehicule și de a folosi utilaje
Injecția cu aflibercept are o influență minoră asupra capacității de a conduce vehicule și de a folosi utilaje din cauza tulburărilor vizuale temporare asociate cu administrarea injecțiilor sau examinarea oculară. Pacienții nu trebuie să conducă vehicule sau să folosească utilaje până când funcția lor vizuală nu s-a restabilit suficient.
4.8 Reacții adverse
Rezumatul profilului de siguranță
Populația de siguranță a fost constituită din 3102 pacienți în opt studii clinice de fază III. Dintre aceștia, 2501 pacienți au fost tratați cu doza recomandată de 2 mg.
Reacțiile adverse oculare grave la nivelul ochiului evaluat în cadrul studiului, asociate cu procedura de injectare au apărut la mai puțin de 1 din 1900 injectări intravitreene cu aflibercept și au inclus: cecitate, endoftalmită, dezlipire de retină, cataractă traumatică, cataractă, hemoragie vitroasă, dezlipire de corp vitros și creștere a presiunii intraoculare (vezi pct. 4.4).
Reacțiile adverse observate cel mai frecvent (apărute la cel puțin 5% dintre pacienții cărora li s-a administrat aflibercept) au fost: hemoragie conjunctivală (25%), hemoragie retiniană (11%), scăderea acuității vizuale (11%), durere oculară (10%), cataractă (8%), creșterea presiunii intraoculare (8%), dezlipire de corp vitros (7%) și flocoanele intravitreene (7%).
Rezumatul reacțiilor adverse – lista sub formă de tabel
Datele privind siguranța, descrise mai jos, includ toate reacțiile adverse provenite din cele opt studii clinice de fază III în indicațiile DMLV forma umedă, OVCR, ORVR, EMD și NVC miopică, cu o posibilitate rezonabilă de cauzalitate legată de procedura de injectare sau de medicament.
Reacțiile adverse sunt enumerate în funcție de clasificarea pe aparate, sisteme și organe, utilizând următoarea convenție:
Foarte frecvente (≥1/10), frecvente (≥1/100 și <1/10), mai puțin frecvente (≥1/1000 și <1/100), rare (≥1/10000 și <1/1000), cu frecvență necunoscută (care nu poate fi estimată din datele disponibile).
În cadrul fiecărei grupe de frecvență, reacțiile adverse la medicament sunt prezentate în ordinea descrescătoare a gravității.
Tabelul 1: Toate reacțiile adverse la medicament asociate tratamentului, raportate la pacienți în studii clinice de fază III (date cumulate din studiile clinice de fază III pentru indicațiile DMLV forma umedă, OVCR, ORVR, EMD și NVC miopică) sau în timpul supravegherii după punerea pe piață
| Clasificarea pe aparate, sisteme și organe | Frecvența | Reacție adversă |
|---|---|---|
| Tulburări ale sistemului imunitar | Mai puțin frecvente | Hipersensibilitate*** |
| Tulburări oculare | Foarte frecvente | Scăderea acuității vizuale, Hemoragie retiniană, Hemoragie conjunctivală, Durere oculară |
| Frecvente | Ruptură a epiteliului retinian pigmentar*, Dezlipirea epiteliului retinian pigmentar, Degenerare retiniană, Hemoragie vitroasă, Cataractă, Cataractă corticală, Cataractă nucleară, Cataractă subcapsulară, Eroziune corneeană, Abraziune corneană, Creşterea presiunii intraoculare, Vedere înceţoşată, Flocoane intravitroase, Dezlipire de corp vitros, Durere la locul de injecţie, Senzaţie de corpi străini la nivel ocular, Hiperlacrimaţie, Edem palpebral, Hemoragie la locul de injecţie, Keratită punctată, Hiperemie conjunctivală, Hiperemie oculară. | |
| Mai puțin frecvente | Endoftalmită**, Dezlipire de retină, Ruptură de retină, Irită, Uveită, Iridocciclită, Opacități lenticulare, Defecte ale epiteliului cornean, Iritație la locul de injecție, Senzație anormală în ochi, Blefarită, Congestie la nivelul camerei anterioare, Edem cornean | |
| Rare | Cecitate, Cataractă traumatică, Vitrită, Hipopion | |
| Cu frecvență necunoscută | Sclerită**** |
* Tulburări cunoscute a fi asociate cu DMLV forma umedă. Observate numai în studiile efectuate la pacienții cu DMLV forma umedă.
** Endoftalmite de cultură pozitivă și cultură negativă
*** În timpul perioadei de după punerea pe piață, raportările de hipersensibilitate au inclus erupții cutanate tranzitorii, prurit, urticarie, și cazuri izolate de reacții anafilactice severe/reacții anafilactoide. **** Din raportările post-marketing
Descrierea reacțiilor adverse selectate
Într-un studiu clinic de fază III efectuat la pacienții cu DMLV forma umedă a fost observată o incidență crescută a hemoragiilor conjunctivale la pacienții tratați cu medicamente antiagregante. Această incidență crescută a fost comparabilă cu cea apărută la pacienții tratați cu ranibizumab și aflibercept.
Evenimentele arteriale tromboembolice (EAT) sunt evenimente adverse potențial corelate cu inhibiția sistemică a VEGF. Există un risc teoretic de evenimente arteriale tromboembolice, inclusiv accident vascular cerebral și infarct miocardic, în urma administrării intravitreene de inhibitori ai VEGF.
A fost observată o rată scăzută a incidenței evenimentelor tromboembolice arteriale în studiile clinice cu aflibercept la pacienții cu DMLV, EMD, OVR și NVC miopică. Referitor la toate indicațiile, nu a fost observată nicio diferență notabilă între grupurile tratate cu aflibercept și grupurile comparator respective. Similar tuturor proteinelor terapeutice, există un risc potențial de imunogenitate în cazul administrării afliberceptului.
Raportarea reacțiilor adverse suspectate
Raportarea reacțiilor adverse suspectate după autorizarea medicamentului este importantă. Acest lucru permite monitorizarea continuă a raportului beneficiu/risc al medicamentului. Profesioniștii din domeniul
| sistemului | național de |
|---|
raportare, astfel cum este menționat în Anexa V.
4.9 Supradozaj
În studiile clinice s-au utilizat doze de până la 4 mg la intervale lunare și au apărut cazuri izolate de supradozaj la doze de 8 mg.
Supradozajul cu un volum crescut de soluție injectabilă poate crește presiunea intraoculară. Prin urmare, în cazul supradozajului trebuie monitorizată presiunea intraoculară și trebuie inițiat tratamentul adecvat de către medicul curant, dacă acest lucru este considerat necesar (vezi pct. 6.6).
5. PROPRIETĂȚI FARMACOLOGICE
5.1 Proprietăți farmacodinamice
Grupa farmacoterapeutică: Medicamente oftalmologice / Medicamente antineovascularizație Codul ATC: S01LA05
| MYNZEPLI | este un medicament biosimilar. Informații detaliate sunt disponibile pe site-ul Agenției | |
| Europene pentru Medicamente https://www.ema.europa.eu | ||
|---|---|---|
Aflibercept este o proteină recombinantă de fuziune, formată din porțiuni ale domeniilor extracelulare ale receptorilor 1 și 2 ai VEGF uman, fuzionate cu porțiunea Fc a IgG1 umane.
Aflibercept este obținut prin tehnologie ADN recombinantă în celule ovariene de hamster chinezesc (CHO) K1.
Aflibercept acționează ca un receptor capcană, solubil, care se leagă de VEGF-A și de factorul placentar de creștere (PlGF), cu afinitate superioară receptorilor naturali ai acestora și astfel poate inhiba legarea și activarea acestor receptori înrudiți ai VEGF.
Mecanism de acțiune
Factorul A endotelial de creștere vasculară (VEGF-A) și factorul placentar de creștere (PlGF) sunt membri ai familiei VEGF a factorilor angiogenici, care pot acționa ca factori puternici mitogeni, chemotactici și de permeabilitate vasculară pentru celulele endoteliale. VEGF acționează prin intermediul a doi receptori ai tirozin kinazelor, VEGFR-1 și VEGFR-2, prezenți pe suprafața celulelor endoteliale. PlGF se leagă numai de VEGFR-1, prezent de asemenea pe suprafața leucocitelor.
Activarea în exces a acestor receptori de către VEGF-A poate determina apariția neovascularizației patologice și creșterea permeabilității vasculare. În aceste procese, PlGF poate acționa sinergic cu VEGF-A și este cunoscut, de asemenea, ca promotor al infiltrației leucocitare și inflamației vasculare.
Efecte farmacodinamice
DMLV forma umedă
DMLV forma umedă se caracterizează prin apariția neovascularizației patologice coroidale (NVC). Scurgerea sângelui și lichidelor de la nivelul NVC poate provoca îngroșare retiniană sau edem retinian și/sau hemoragie sub-/intraretiniană, ceea ce duce la pierderea acuității vizuale.
La pacienții tratați cu aflibercept (o injecție administrată lunar, timp de trei luni consecutiv, urmată de o injecție la intervale de 2 luni), grosimea centrală a retinei [GCR] a scăzut curând după inițierea tratamentului și dimensiunea medie a leziunii NVC a scăzut, ceea ce confirmă rezultatele observate în cazul administrării ranibizumabului lunar în doză de 0,5 mg.
În studiul clinic VIEW1 s-au evidențiat scăderi medii ale GCR la tomografia în coerență optică (TCO) a scăderii medii ale îngroșării retinei (-130 și -129 microni în săptămâna 52 pentru grupele de studiu la care s-a administrat aflibercept 2 mg la intervale de 2 luni și, respectiv, ranibizumab 0,5 mg administrat lunar). De asemenea, la momentul săptămânii 52 în cadrul studiului clinic VIEW2, TCO a evidențiat scăderi medii ale GCR (-149 și -139 microni pentru grupele de studiu la care s-au administrat aflibercept 2 mg la intervale de 2 luni și, respectiv, ranibizumab 0,5 mg administrat lunar). Scăderea dimensiunii NVC și scăderea GCR au fost în general menținute în al 2-lea an al studiilor.
Studiul clinic ALTAIR, efectuat la pacienți japonezi, cu DMLV forma umedă, cărora nu li se administrase tratament anterior, arată rezultate similare cu ale studiilor VIEW, folosind 3 injectări lunare inițiale, de 2 mg aflibercept, urmate de o injectare după alte 2 luni, iar apoi continuat cu un regim de tip „tratament și extindere” cu intervale de tratament variabile (ajustări de 2 sau 4 săptămâni) până la un interval de maximum 16 săptămâni, conform criteriilor specificate anterior.
În săptămâna 52, au existat scăderi medii ale grosimii centrale a retinei (GCR) la TCO, de
-134,4 și -126,1 microni pentru grupul cu ajustare de 2 săptămâni și respectiv grupul cu ajustare de 4 săptămâni. Proporția pacienților fără fluid la TCO în săptămâna 52 a fost de 68,3% și 69,1% în grupurile cu ajustare de 2 și 4 săptămâni. În general, scăderea GCR s-a păstrat în ambele brațe ale studiului ALTAIR în al doilea an.
Studiul clinic ARIES a fost proiectat pentru explorarea non-inferiorității unui regim de doze de tipul tratament și extindere cu aflibercept 2 mg inițiat imediat după administrarea inițială a 3 injectări lunare și o injectare suplimentară după 2 luni versus un regim de doze de tipul tratament și extindere inițiat la un an după tratament. Pentru pacienții care aveau nevoie de doze mai frecvente decât Q8, cel puțin o dată pe parcursul studiului, GCR a rămas mărită, dar scăderea medie a GCR de la nivelul de bază la săptămâna 104 a fost -160,4 microni, similară cu cea a pacienților tratați la Q8 sau intervale mai puțin frecvente.
Edem macular secundar OVCR și ORVR
În OVCR și ORVR apare ischemia retiniană, aceasta semnalând eliberarea VEGF care, la rândul său, destabilizează joncțiunile compacte și stimulează proliferarea celulelor endoteliale. Reglarea ascendentă a VEGF este asociată cu ruperea barierei hemato-retiniene, permeabilitate vasculară crescută, edem retinian și complicații ale neovascularizației.
La pacienții tratați cu o injecție de aflibercept 2 mg lunar, timp de 6 luni consecutiv, s-a observat un răspuns consistent, rapid și intens în ceea ce privește morfologia (măsurată prin îmbunătățirea medie a GCR). În săptămâna 24, reducerea GCR a fost statistic superioară, față de grupul de control, în toate cele 3 studii clinice (COPERNICUS în OVCR: -457 vs. -145 microni; GALILEO în OVCR: -449 vs. -169 microni; VIBRANT în ORVR: -280 vs. -128 microni). Această reducere a GCR față de momentul inițial s-a menținut până la sfârșitul fiecărui studiu clinic, săptămâna 100 în studiul clinic COPERNICUS, săptămâna 76 în studiul clinic GALILEO și săptămâna 52 în studiul clinic VIBRANT.
Edem macular diabetic
Edemul macular diabetic este o consecință a retinopatiei diabetice și se caracterizează prin creșterea permeabilității vasculare și deteriorarea capilarelor retiniene, care poate duce la pierderea acuității vizuale.
La pacienții tratați cu aflibercept, cea mai mare parte dintre aceștia având diagnostic de diabet zaharat de tip 2, s-a observat un răspuns rapid și intens în ceea ce privește morfologia (GCR, scorul DRSS).
În studiile clinice VIVIDDME și VISTADME, scăderile medii semnificativ statistic mai mari ale GCR față de valoarea inițială în săptămâna 52 au fost observate la pacienții tratați cu aflibercept comparativ cu pacienții din grupul de control cu laser, fiind de -192,4 microni și -183,1 microni pentru grupurile tratate cu aflibercept 2Q8 și, respectiv, -66,2 microni și -73,3 microni pentru grupurile de control. În săptămâna 100, scăderea s-a menținut, cu -195,8 microni și -191,1 microni pentru grupurile tratate cu aflibercept 2Q8 și respectiv cu -85,7 și -83,9 microni pentru grupurile de control, în studiile VIVIDDME și VISTADME.
În studiile VIVIDDME și VISTADME, o îmbunătățire ≥ 2 trepte a scorului DRSS (Scorul severității retinopatiei diabetice) a fost evaluată în manieră prespecificată. Scorul DRSS a putut fi clasificat pe grade la 73,7% dintre pacienții din studiul VIVIDDME și la 98,3% dintre pacienții din studiul VISTADME. În săptămâna 52, la 27,7% și 29,1% dintre pacienții din grupurile tratate cu aflibercept 2Q8 și la 7,5% și 14,3% dintre pacienții din grupurile de control s-a înregistrat o îmbunătățire ≥ 2 trepte a scorului DRSS. În săptămâna 100, procentele respective au fost de 32,6% și 37,1% dintre pacienții din grupurile tratate cu aflibercept 2Q8 și 8,2% și 15,6% dintre pacienții din grupurile de control.
Studiul clinic VIOLET a comparat trei regimuri diferite de dozare a aflibercept 2 mg pentru tratamentul EMD după cel puțin un an de tratament la intervale fixe, când tratamentul a fost inițiat cu 5 doze lunare consecutive urmate de doze la fiecare 2 luni. În săptămâna 52 și săptămâna 100 ale studiului, adică al doilea și al treilea an de tratament, media schimbărilor GCR era similară din punct de vedere clinic pentru “tratament și extindere” (2T&E), pro re nata (2PRN) și 2Q8, respectiv cu variații de -2,1, 2,2 și -18,8 microni în săptămâna 52 și variații de 2,3, -13,9 și - 15,5 microni în săptămâna 100.
Neovascularizație coroidală miopică
Neovascularizația coroidală miopică (NVC miopică) este o cauză frecventă a pierderii vederii la adulții cu miopie patologică. Aceasta apare ca mecanism de vindecare a leziunilor, în urma ruperii membranei Bruch și reprezintă cel mai dăunător eveniment pentru vedere în miopia patologică.
La pacienții tratați cu aflibercept în studiul clinic MYRROR (o injecție administrată la începutul terapiei, fiind administrate injecții suplimentare în cazul persistenței sau al recurenței bolii) GCR s-a diminuat la scurt timp după inițierea tratamentului fiind în favoarea tratamentului cu aflibercept în săptămâna 24 (-79 microni pentru grupul tratat cu aflibercept 2 mg și, respectiv, -4 microni pentru grupul de control), care s-a menținut până în săptămâna 48. În plus, leziunea NVC medie s-a redus.
Eficacitate și siguranță clinică
DMLV forma umedă
Siguranța și eficacitatea clinică a afliberceptului au fost evaluate în două studii randomizate, multicentrice, cu dublă mascare a formei farmaceutice, controlate activ, la pacienți cu DMLV forma umedă (VIEW1 și
VIEW2) cu un număr total de 2412 pacienți tratați și evaluați în vederea stabilirii eficacității (1817 pacienți la care s-a administrat aflibercept).Vârsta pacienților a variat de la 49 ani la 99 ani, cu o medie de 76 ani. În aceste studii clinice, aproximativ 89% (1,616/1,817) dintre pacienții randomizați la tratamentul cu aflibercept aveau vârsta de 65 ani sau mai mult, iar aproximativ 63% (1,139/1,817) aveau vârsta de 75 ani sau mai mult. În cadrul fiecărui studiu, pacienții au fost repartizați randomizat, în raport de 1:1:1:1, la unul din cele 4 regimuri de dozaj:
1) Aflibercept administrat în doză de 2 mg la intervale de 8 săptămâni, după administrarea a 3 doze inițiale la intervale lunare (aflibercept 2Q8);
2) Aflibercept administrat în doză de 2 mg la intervale de 4 săptămâni (aflibercept 2Q4);
3) Aflibercept administrat în doză de 0,5 mg la intervale de 4 săptămâni (aflibercept 0,5Q4); și
4) ranibizumab administrat în doză de 0,5 mg la intervale de 4 săptămâni (ranibizumab 0,5Q4).
În al doilea an al studiilor, pacienților li s-a administrat în continuare doza la care au fost inițial repartizați randomizat, dar cu un program modificat de administrare a dozelor, pe baza evaluării rezultatelor vizuale și anatomice, cu un interval maxim de dozaj de 12 săptămâni, definit de protocol.
În ambele studii, obiectivul principal al eficacității a fost procentul pacienților din grupele de studiu, care șiau menținut acuitatea vizuală, de exemplu pierderea a mai puțin de 15 litere din acuitatea vizuală în săptămâna 52, de la momentul inițial.
În studiul clinic VIEW1, în săptămâna 52, 95,1% dintre pacienții din grupul tratat cu aflibercept 2Q8 au menținut acuitatea vizuală, comparativ cu 94,4% dintre pacienții în grupul căruia s-a administrat ranibizumab 0,5Q4. În studiul clinic VIEW2, în săptămâna 52, 95,6% dintre pacienții din grupul tratat cu aflibercept 2Q8 și-au menținut acuitatea vizuală, comparativ cu 94,4% dintre pacienții în grupul căruia s-a administrat ranibizumab 0,5Q4. În ambele studii, tratamentul cu aflibercept s-a dovedit a fi non-inferior și echivalent din punct de vedere clinic cu grupul căruia s-a administrat ranibizumab 0,5Q4.
Rezultatele detaliate provenite din analiza centralizată a ambelor studii sunt prezentate în Tabelul 3 și Figura 1 de mai jos.
Tabelul 2: Rezultatele privind eficacitatea în săptămâna 52 (analiză primară) și în săptămâna 96; date combinate provenite din studiile VIEW1 și VIEW2B)
| Rezultat privind eficacitatea | Aflibercept 2Q8 E) (aflibercept 2 mg la intervale de 8 săptămâni, după administrarea a 3 doze inițiale la intervale lunare) (N = 607) | Ranibizumab 0,5Q4 (ranibizumab 0,5 mg la intervale de 4 săptămâni) (N = 595) | ||
| Săptămâna 52 | Săptămâna 96 | Săptămâna 52 | Săptămâna 96 | |
| Numărul mediu de injecții | 7,6 | 11,2 | 12,3 | 16,5 |
| Numărul mediu de injecții din săptămâna 52 până în săptămâna 96 | 4,2 | 4,7 | ||
Procentul pacienților cu < 15 litere pierdute față de momentul inițial SPPA) | 95,33%B) | 92,42% | 94,42%B) | 91,60% |
| DiferențăC) (IÎ95%)D) | 0,9% (-1,7; 3,5)F) | 0,8% (-2,3; 3,8)F) | ||
| Modificarea medie a AVOC măsurată prin scorul literelor ETDRSA) față de momentul inițial | 8,40 | 7,62 | 8,74 | 7,89 |
| Diferența în media celor mai | ||||
|---|---|---|---|---|
mici pătrate A) (litere ETDRS)C) (IÎ95%)D) | -0,32 (-1,87; 1,23) | -0,25 (-1,98; 1,49) | ||
Procentul pacienților cu ≥ 15 litere câștigate față de momentul inițial | 30,97% | 33,44% | 32,44% | 31,60% |
DiferențăC) IÎ(95%)D) | -1,5% (-6,8; 3,8) | 1,8% (-3,5; 7,1) | ||
A) AVOC: Acuitatea vizuală optim corectată
ETDRS: Early Treatment Diabetic Retinopathy Study (Studiul retinopatiei diabetice cu tratament precoce) LS: Media celor mai mici pătrate derivată din ANCOVA
SPP: Set Per Protocol (set pentru fiecare protocol)
B) Setul complet de analiză (FAS), Extrapolarea în sens longitudinal a ultimelor date observate (LOCF) pentru toate analizele, cu excepția procentului de pacienți la care acuitatea vizuală a fost menținută în săptămâna 52, reprezentând setul pentru fiecare protocol (PPS)
C) Diferența este reprezentată de valoarea grupului cu aflibercept minus valoarea grupului cu ranibizumab. O valoare pozitivă favorizează aflibercept.
D) Intervalul de încredere (IÎ) calculat prin aproximare normală
E) După inițierea tratamentului cu doze administrate la intervale de trei luni
F) Un interval de încredere complet superior valorii de -10% indică non-inferioritatea aflibercept față de ranibizumab
Figura 1 Modificarea medie a acuității vizuale de la momentul inițial până în săptămâna 96 pentru datele centralizate provenite din studiile View1 și View2
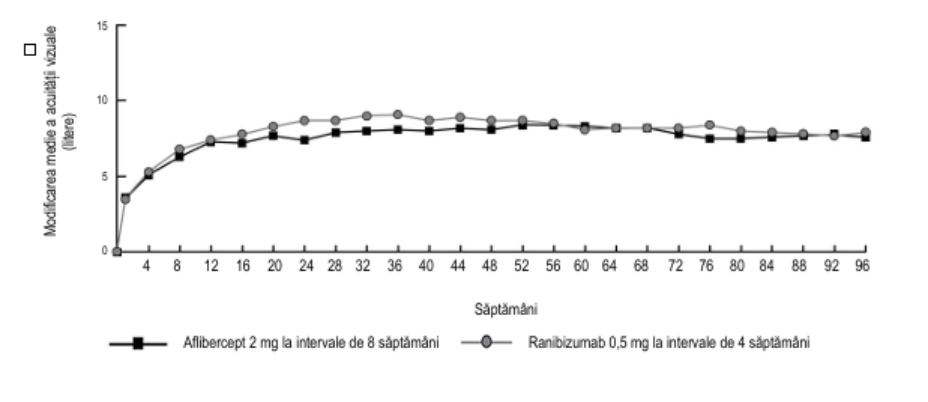
În analiza datelor centralizate provenite din studiile VIEW1 și VIEW2, MYNZEPLI a demonstrat modificări semnificative din punct de vedere clinic față de momentul inițial, conform Chestionarului privind funcția vizuală, al Institutului Național pentru Afecțiuni Oculare (NEI VFQ-25), fără diferențe semnificative clinic față de ranibizumab. Importanța acestor modificări a fost similară celor observate în studiile publicate, corespunzând unui câștig de 15 litere din acuitatea vizuală optim corectată (AVOC).
În al doilea an al studiilor, eficacitatea s-a menținut, în general, conform ultimei evaluări din săptămâna 96, și 2-4% dintre pacienți au necesitat administrarea lunară a tuturor injecțiilor, iar 1/3 dintre pacienți au necesitat cel puțin o injecție la un interval de tratament de numai o lună.
Scăderile medii ale zonei NVC au fost evidente la toate grupele de tratament în ambele studii.
Rezultatele privind eficacitatea în toate subgrupele care au putut fi evaluate (de exemplu vârstă, sex, rasă, acuitate vizuală la momentul inițial, tipul leziunii, mărimea leziunii) în fiecare studiu și în analiza combinată au confirmat rezultatele în populația generală.
ALTAIR a fost un studiu multicentric de 96 de săptpmâni, randomizat, deschis, efectuat la 247 pacienți japonezi cu DMLV forma umedă, naivi la tratament, dezvoltat pentru a evalua eficacitatea și siguranța afliberceptului după două intervale (2 săptămâni și 4 săptămâni) de ajustare a unui regim de tip „tratament și extindere”.
Tuturor pacienților li s-au administrat doze lunare de aflibercept de 2 mg , timp de 3 luni, urmate de o injectare după un interval de 2 luni. În săptămâna 16, pacienții au fost randomizați 1:1 în două grupuri de tratament: 1) aflibercept „tratament și extindere” cu ajustări la 2 săptămâni și 2) aflibercept „tratament și extindere” cu ajustări la 4 săptămâni. Decizia de a extinde sau a scurta intervalele de tratament a fost luată în funcție de criteriile vizuale și/sau anatomice definite de protocol cu un interval de tratament de maximum 16 săptămâni pentru ambele grupuri.
Criteriul principal de evaluare a eficacității a fost schimbarea medie a AVOC de la nivelul de bază la săptămâna 52. Criteriile secundare de evaluare a eficacității au fost reprezentate de proporția de pacienți care nu au pierdut ≥15 litere și proporția de pacienți care au câștigat cel puțin 15 litere din AVOC de la nivelul de bază la săptămâna 52.
În săptămâna 52, pacienții din brațul de „tratament și extindere” cu ajustări la 2 săptămâni au obținut un câștig mediu de 9,0 litere din nivelul de bază, comparativ cu 8,4 litere pentru cei din grupul cu ajustare la 4 săptămâni [LS diferența medie în litere (95% IÎ): -0,4 (-3,8, 3,0), ANCOVA]. Proporția de pacienți care nu au pierdut ≥15 litere în cele două brațe de tratament a fost similară (96,7% la grupul cu ajustare la 2 săptămâni și 95,9% la 4 săptămâni). Proporția de pacienți ce au avut un câștig ≥15 litere în săptămâna 52 a fost 32,5% în grupul cu ajustare la 2 săptămâni și 30,9% în grupul cu ajustare la 4 săptămâni. Proporția de pacienți la care s-a extins intervalul de tratament la 12 săptămâni sau mai mult a fost 42,3% în grupul cu ajustare la 2 săptămâni și 49,6% în grupul cu ajustare la 4 săptămâni. Suplimentar, în grupul cu ajustare la 4 săptămâni, la 40,7% din pacienți s-a extins intervalul de tratament la 16 săptămâni. La ultima vizită până la săptămâna 52, 56,8% și 57,8% dintre pacienții din grupurile cu ajustare la 2 și respectiv 4 săptămâni au avut următoarea injectare programată la un interval de 12 săptămâni sau mai mare.
În al doilea an de studiu, în general eficacitatea s-a păstrat până la și incluzând ultima evaluare din săptămâna 96, cu un câștig mediu față de nivelul de bază de 7,6 litere în grupul cu ajustare la 2 săptămâni și de 6,1 litere în grupul cu ajustare la 4 săptămâni. Proporția de pacienți la care s-a extins intervalul de tratament la 12 săptămâni sau peste acest interval a fost de 56,9% în grupul cu ajustare la 2 săptămâni și de 60,2% în grupul cu ajustare la 4 săptămâni. La ultima vizită, înainte de săptămâna 96, la 64,9% și 61,2% dintre pacienții din grupul cu ajustare la 2 și respectiv 4 săptămâni a fost planificată următoarea injecție la un interval de 12 săptămâni sau mai mare. În timpul celui de-al doilea an de tratament pacienții din cele două grupuri cu ajustare la 2 și respectiv 4 săptămâni li s-a administrat în medie 3,6 și, respectiv 3,7 injecții. Pentru perioada de 2 ani de tratament, pacienților li s-au administrat o medie de 10,4 injecții.
Profilurile de siguranță oculară și sistemică au fost similare cu siguranța observată în studiile pivot VIEW1 și VIEW2.
ARIES a fost un studiu cu o durată de 104 săptămâni, multicentric, randomizat, în regim deschis, controlat cu comparator activ efectuat la 269 pacienți naivi la tratament pentru DMLV forma umedă, proiectat pentru a evalua non-inferioritatea în termeni de eficacitate precum și siguranță a unui regim de doze de tipul tratament și extindere inițiat după 3 doze lunare consecutive urmate de o extindere la un interval de tratament de 2 luni versus regimul de doze tip tratament și extindere inițiat după primul an de tratament.
Studiul ARIES a explorat de asemenea și procentul de pacienți care au necesitat tratament mai frecvent de 8 săptămâni pe baza deciziei investigatorului. Din 269 pacienți, 62 pacienți au primit doze mai frecvente cel puțin o dată pe parcursul studiului. Acești pacienți au rămas în studiu și au primit tratament în acord cu evaluarea clinică cea mai bună a investigatorului, dar nu mai frecvent de 4 săptămâni și intervalele de tratament au putut fi extinse din nou ulterior. Intervalul mediu de tratament după decizia de a trata mai frecvent a fost de 6,1 săptămâni. În săptămâna 104 AVOC a fost mai mică la pacienții care au avut nevoie de tratament intensiv cel puțin o dată pe parcursul studiului, comparativ cu pacienții care nu au au avut nevoie, iar schimbarea medie în AVOC la finalul studiului față de nivelul de bază a fost de +2,3 ± 15,6 litere. Printre pacienții tratați mai frecvent, 85,5% și-au menținut vederea, de exemplu au pierdut mai puțin de 15 litere și 19,4% au câștigat 15 litere sau mai multe. Profilul de siguranță pentru pacienții tratați mai frecvent de 8 săptămâni a fost comparabil cu datele de siguranță din studiile VIEW 1 și VIEW 2.
Edem macular secundar OVCR
Siguranța și eficacitatea clinică a afliberceptului au fost evaluate în două studii randomizate, multicentrice, cu dublă mascare a formei farmaceutice, controlate cu tratament fictiv, la pacienți cu edem macular secundar OVCR (COPERNICUS și GALILEO cu un număr total de 358 pacienți care au fost tratați și evaluați în vederea stabilirii eficacității (217 cu aflibercept). Vârsta pacienților a variat între 22 ani și 89 ani, cu o medie de 64 ani. În studiile efectuate la pacienți cu OVCR, aproximativ 52% (112/217) dintre pacienții randomizați la tratamentul cu MYNZEPLI aveau vârsta de 65 ani sau mai mult, și aproximativ 18% (38/217) aveau vârsta de 75 ani sau mai mult. În ambele studii, pacienții au fost repartizați randomizat, în raport de 3:2, fie în grupul cu aflibercept 2 mg administrat la intervale de 4 săptămâni (2Q4), fie în grupul de control, pentru a li se administra injecții cu tratament fictiv, la intervale de 4 săptămâni, pentru un total de 6 injecții.
După administrarea unei injecții lunar, timp de 6 luni consecutiv, pacienților li s-a administrat tratament numai dacă au întrunit criteriile specificate în prealabil pentru repetarea tratamentului, cu excepția pacienților din grupul de control din cadrul studiului clinic GALILEO, care au continuat administrarea de tratament fictiv (control față de control) până în săptămâna 52. Începând din acest moment, tuturor pacienților li s-a administrat tratament dacă au întrunit criteriile specificate în prealabil.
În ambele studii, obiectivul principal al eficacității a fost procentul pacienților care a câștigat cel puțin 15 litere din AVOC în săptămâna 24, comparativ cu momentul inițial. A doua variabilă de eficacitate secundară a fost modificarea acuității vizuale în săptămâna 24 comparativ cu momentul inițial.
Diferența între grupurile de tratament a fost favorabilă pentru aflibercept într-un mod semnificativ statistic, în ambele studii. Îmbunătățirea maximă a acuității vizuale s-a realizat la 3 luni cu stabilizarea ulterioară asupra acuității vizuale și asupra GCR până la 6 luni. Diferența semnificativă statistic a fost menținută până în săptămâna 52.
Rezultatele detaliate din analiza ambelor studii sunt prezentate în Tabelul 3 și Figura 2 de mai jos.
Tabelul 3: Rezultatele privind eficacitatea în săptămâna 24, săptămâna 52 și săptămâna 76/100 (Set complet de analiză cu LOCFC)) în studiile COPERNICUS și GALILEO
| Rezultate privind eficacitatea | Studiul COPERNICUS | Studiul GALILEO | ||||||||||
| 24 săptămâni | 52 săptămâni | 100 săptămâni | 24 săptămâni | 52 săptămâni | 76 săptămâni | |||||||
| Aflibercept 2 mg Q4 (N = 114) | Control (N= 73) | Aflibercept 2 mg (N = 114) | Control E) (N =73) | Aflibercept F) 2 mg (N = 114) | Control E,F) (N=73) | Aflibercept 2 mg Q4 (N = 103) | Control (N = 68) | Aflibercepti 2 mg (N = 103) | Control (N = 68) | Aflibercept G) 2 mg (N = 103) | Control G) (N = 68) | |
| Procentul pacienților care au câștigat ≥15 litere din AVOCC) față de momentul inițial | 56% | 12% | 55% | 30% | 49,1% | 23.3% | 60% | 22% | 60% | 32% | 57,3% | 29.4% |
| Diferența ponderatăA,B,E) (IÎ 95%) valoare-p | 44,8% (33,0; 6,6) p < 0,0001 | 25,9% (11,8; 40,1) p = 0,0006 | 26,7% (13,1; 40,3) p=0,0003 | 38,3% (24,4; 52,1) p < 0,0001 | 27,9% (13,0; 42,7) p = 0,0004 | 28,0% (13,3; 42,6) p=0,0004 | ||||||
Modificarea medie în AVOCC) măsurată prin scorul literelor ETDRSC) față de momentul inițial (DS) | 17,3 (12,8) | -4.0 (18.0) | 16,2 (17,4) | 3.8 (17.1) | 13,0 (17,7) | 1.5 (17.7) | 18,0 (12,2) | 3.3 (14.1) | 16,9 (14,8) | 3.8 (18.1) | 13,7 (17,8) | 6.2 (17.7) |
| Diferența în valoarea A,C,D,E) medie a LS (IÎ 95%) valoarea-p | 21,7 (17,4; 6,0) p < 0,0001 | 12,7 (7,7; 17,7) p < 0,0001 | 11,8 (6,7; 17,0) p < 0,0001 | 14.7 (10,8; 18,7) p < 0,0001 | 13,2 (8,2; 18,2) p < 0,0001 | 7,6 (2,1; 13,1) p=0,0070 | ||||||
A) Diferența este reprezentată de tratamentul cu aflibercept 2 mg administrat o dată la 4 săptămâni minus tratament de control B) Diferența și intervalul de încredere (IÎ) sunt calculate utilizând testul Cochran-Mantel-Haenszel (CMH) ajustat în funcție de regiune (America față de restul lumii pentru studiul clinic COPERNICUS și Europa față de Asia/Pacific pentru studiul clinic GALILEO) și AVOC la momentul inițial (> 20/200 and ≤ 20/200)
C) AVOC: Acuitatea vizuală optim corectată
ETDRS: Early Treatment Diabetic Retinopathy Study (Studiul retinopatiei diabetice cu tratament precoce) LOCF: Extrapolarea în sens longitudinal a ultimelor date observate
DS: Deviație standard
LS: Media celor mai mici pătrate derivată din ANCOVA
D) LS diferența în media celor mai mici pătrate și intervalul de încredere (IÎ) pe baza modelului ANCOVA cu factori de tipul: grup de tratament, regiune
46
(America față de restul lumii pentru studiul clinic COPERNICUS și Europa față de Asia/Pacific pentru studiul clinic GALILEO) și AVOC la momentul inițial (> 20/200 și ≤ 20/200)
E) În studiul clinic COPERNICUS, pacienților din grupul de control li s-a putut administra aflibercept la nevoie, la intervale de 4 săptămâni, din săptămâna 24 până în săptămâna 52; pacienții efectuau vizite la interval de 4 săptămâni
F) În studiul clinic COPERNICUS, atât pacienților din grupul de control cât și cei cărora li s-a administrat aflibercept 2 mg, li s-a putut administra aflibercept 2 mg la nevoie, la intervale de 4 săptămâni, din săptămâna 52 până în săptămâna 96; pacienții efectuau vizite trimestriale obligatorii, dar era posibil să fie consultați cu o frecvență de 4 săptămâni, în cazul în care era necesar.
G) În studiul clinic GALILEO, atât pacienților din grupul de control cât și cei cărora li s-a administrat MYNZEPLI 2 mg, li s-a putut administra MYNZEPLI 2 mg la nevoie, la intervale de 8 săptămâni, din săptămâna 52 până în săptămâna 68; pacienții efectuau vizite la interval de 8 săptămâni.
47
Figura 2: Modificarea medie de la momentul inițial în săptămâna 76/100 în ceea ce privește acuitatea vizuală în funcție de grupul de tratament pentru studiile COPERNICUS și GALILEO (Set complet de
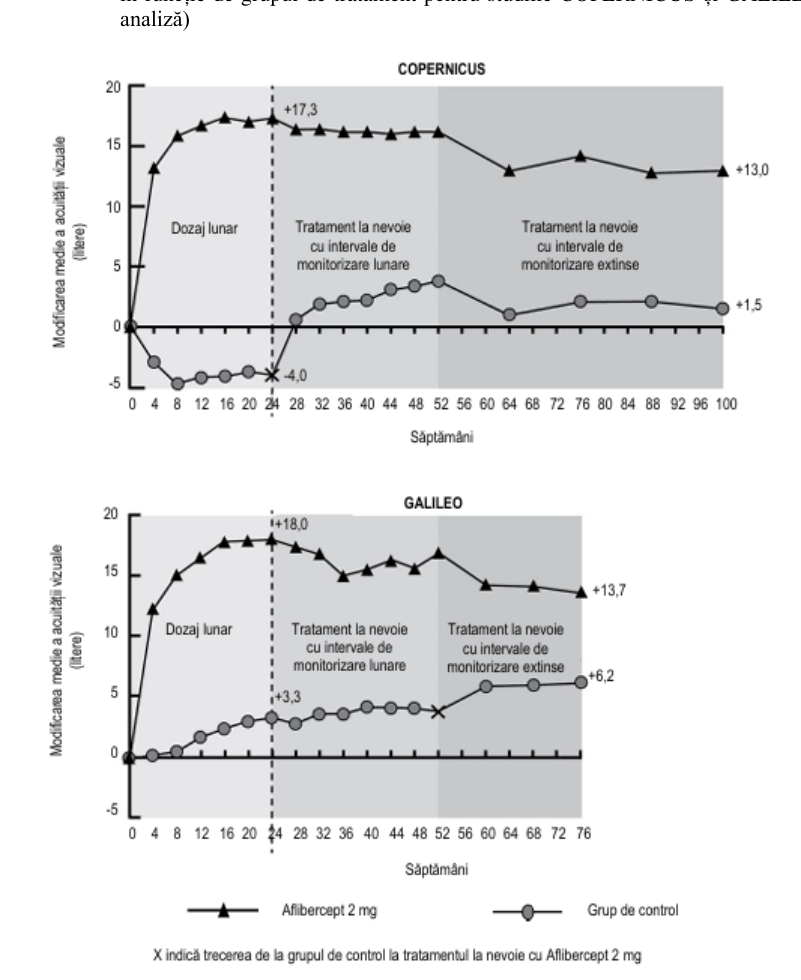
În studiul clinic GALILEO, un procent de 86,4% (n=89) din grupul care a primit tratament cu aflibercept și 79,4% (n = 54) din grupul care a primit tratament fictiv au fost injectați pentru OVCR la momentul inițial. În săptămâna 24, procentul a fost de 91,8% (n = 89) în grupul care a primit tratament cu aflibercept și 85,5% (n = 47) în grupul care a primit tratament fictiv. Aceste proporții s-au menținut în săptămâna 76, cu 84,3% (n = 75) în grupul care a primit tratament cu aflibercept și 84,0% (n = 42) în grupul care a primit tratament fictiv.
În studiul clinic COPERNICUS, un procent de 67,5% (n=77) din grupul care a primit tratament cu aflibercept și 68,5% (n = 50) din grupul care a primit tratament fictiv au fost injectați pentru OVCR la momentul inițial. În săptămâna 24, procentul a fost de 87,4% (n = 90) în grupul care a primit tratament cu aflibercept și 58,6% (n = 34) în grupul care a primit tratament fictiv. Aceste proporții s-au menținut în săptămâna 100, 76,8% (n = 76) în grupul care a primit tratament cu aflibercept și 78% (n = 39) în grupul care a primit tratament fictiv. Pacienții din grupul care a primit tratament fictiv au fost eligibili pentru a primi aflibercept începând cu săptămâna 24.
Efectul benefic al tratamentului cu aflibercept asupra funcției vizuale a fost similar la grupurile inițiale de pacienți perfuzați și ne-perfuzați. Rezultatele privind eficacitatea în alte subgrupuri, care au putut fi evaluate în fiecare studiu (de exemplu: vârstă, sex, rasă, acuitate vizuală la momentul inițial, durata OVCR), au confirmat rezultatele în populația generală.
În analiza datelor centralizate provenite din studiile clinice GALILEO și COPERNICUS, aflibercept a demonstrat modificări semnificative din punct de vedere clinic față de momentul inițial, conform Chestionarului privind funcția vizuală, al Institutului Național pentru Afecțiuni Oculare (NEI VFQ- 25). Importanța acestor modificări a fost similară celor observate în studiile publicate, corespunzând unui câștig de 15 litere din acuitatea vizuală optim corectată (AVOC).
Edem macular secundar ORVR
Siguranța și eficacitatea aflibercept au fost evaluate printr-un studiu randomizat, multicentric, cu dublu orb, controlat, cu comparator activ, desfășurat la pacienți cu edem macular secundar ORVR (ocluziei de ram venos retinian) (Studiul VIBRANT), inclusiv ocluziei hemiretiniene a venei centrale a retinei. În studiul clinic VIBRANT, un număr total de 181 de pacienți au fost tratați și evaluați privind eficacitatea (la 91 dintre pacienți s-a administrat tratament cu aflibercept). Vârsta pacienților a variat între 42 ani și 94 ani, cu o medie de 65 ani. În cadrul studiului la pacienții cu ORVR, aproximativ 58% (53/91) dintre pacienții randomizați la tratamentul cu aflibercept aveau vârsta de 65 ani sau mai mult, iar aproximativ 23% (21/91) aveau vârsta de 75 ani sau mai mult. În cadrul studiului, pacienții au fost randomizați într-un raport de 1:1, fie pentru a li se administra aflibercept 2 mg la interval de 8 săptămâni - după 1 injecție administrată lunar, timp de 6 luni consecutiv, fie pentru a li se efectua fotocoagulare laser la momentul inițial (grupul de control laser). A fost posibil ca pacienților din grupul de control laser să li se efectueze fotocoagulare laser suplimentară (denumită „tratament laser de salvare“) începând cu săptămâna 12, la un interval minim de 12 săptămâni. Pe baza unor criterii prestabilite pacienții din grupul de tratament laser au primit tratament de salvare cu aflibercept 2 mg începând cu săptămâna 24, administrat la interval de 4 săptămâni, timp de 3 luni consecutiv, urmat de injecții intravitreene la interval de 8 săptămâni.
În studiul clinic VIBRANT, criteriul principal de eficacitate a fost reprezentat de proporția de pacienți care au realizat un câștig de cel puțin 15 litere din AVOC în săptămâna 24 în comparație cu momentul inițial și grupul tratat cu aflibercept a fost superior grupului de control laser.
Un obiectiv secundar de eficacitate a fost modificarea acuității vizuale în săptămâna 24, comparativ cu valoarea inițială, care a fost semnificativ în favoarea afliberceptului, din punct de vedere statistic, în studiul clinic VIBRANT. Evoluția ameliorării vizuale a fost rapidă, iar îmbunătățirea maximă a fost atinsă în luna a-3-a, cu menținerea efectului până în luna 12. În grupul tratat cu laser, la 67 de pacienți li s-a administrat tratament de salvare cu aflibercept începând cu săptămâna 24 (grupul comparator activ/grupul tratat cu aflibercept 2 mg) ceea ce a dus la ameliorarea acuității vizuale cu aproximativ 5 litere din săptămâna 24 până în săptămâna 52.
Rezultate detaliate din analiza studiului clinic VIBRANT sunt prezentate în Tabelul 4 și Figura 3 de mai jos.
Tabelul 4: Rezultatele privind eficacitatea în săptămâna 24 și săptămâna 52 (Set complet de analiză cu LOCFC) în studiul clinic VIBRANT
| Rezultate privind eficacitatea | Studiul VIBRANT | |||
| 24 săptămâni | 52 săptămâni | |||
| Aflibercept 2 mg Q4 (N = 91) | Control Activ (laser) (N = 90) | Aflibercept 2 mg Q8 (N = 91) D) | Control Activ (laser)/ Aflibercept 2 mgE) (N = 90) | |
| Procentul pacienților care au câștigat ≥ 15 litere față de momentul inițial (%) | 52,7% | 26,7% | 57,1% | 41,1% |
| Diferența ponderatăA,B) (IÎ 95%) valoare-p | 26,6% (13,0; 40,1) p=0,0003 | 16,2% (2,0; 30,5) p=0,0296 | ||
| Modificarea medie în AVOC măsurată prin scorul literelor ETDRS față de momentul inițial (DS) | 17,0 (11,9) | 6,9 (12,9) | 17,1 (13,1) | 12,2 (11,9) |
| Diferența în valoarea medie a A,C LS (IÎ 95%) valoare-p | 10,5 (7,1; 14,0) p<0,0001 | 5,2 (1,7; 8,7) p=0,0035F) | ||
A) Diferența este reprezentată de aflibercept 2 mg administrat la interval de 4 săptămâni minus tratament de control activ (laser)
B) Diferența și intervalul de încredere (IÎ) sunt calculate utilizând schema de ponderare Mantel-Haenszel ajustat în funcție de regiune (America față de Japonia) și AVOC la momentul inițial (> 20/200 și ≤ 20/200)
C) LS diferența în media celor mai mici pătrate și intervalul de încredere (IÎ 95%) pe baza modelului ANCOVA cu factori de tipul: grup de tratament, AVOC la momentul inițial (> 20/200 și ≤ 20/200) și regiune (America față de Japonia) ca efecte fixe, și AVOC la momentul inițial ca și co-variantă. D) Din săptămâna 24, intervalul de tratament, al grupului tratat cu aflibercept, a fost extins pentru toți pacienții de la 4 săptămâni la 8 săptămâni în timpul săptămânii 48.
E) Începând cu săptămâna 24, pacienților din grupul tratat cu laser li s-ar putea administra tratament de salvare cu aflibercept, dacă s-a atins cel puțin un criteriu prespecificat de eligibilitate. La un număr total de 67 subiecți din acest grup li s-a administrat tratament cu aflibercept de salvare. Regimul stabilit pentru tratamentul cu aflibercept de salvare a fost de 2 mg la interval de 4 săptămâni, urmate de administrarea unei injecții la interval de 8 săptămâni.
F) Valoare-p nominală
Figura 3: Studiul clinic VIBRANT - Modificarea medie a AVOC măsurată prin scorul literelor ETDRS față de momentul inițial în săptămâna 52
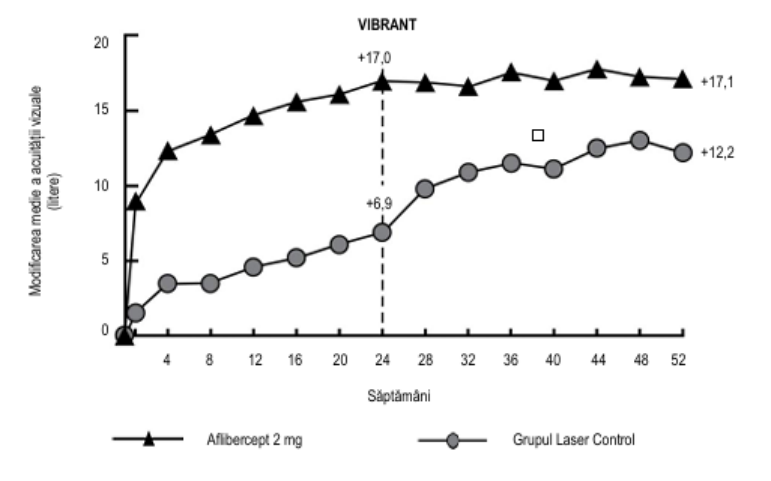
La momentul inițial, proporția pacienților perfuzați în grupul tratat cu aflibercept și grupul tratat cu laser a fost de 60% și, respectiv, 68%. În săptămâna 24, proporția pacienților perfuzați în grupul tratat cu aflibercept și grupul tratat cu laser a fost de 80% și, respectiv, 67%. În grupul tratat cu aflibercept, proporția pacienților perfuzați a fost menținută până la săptămâna 52. În grupul tratat cu laser, în care pacienții au fost eligibili pentru tratamentul cu aflibercept de salvare din săptămâna 24, proporția pacienților perfuzați a crescut la 78% până în săptămâna 52.
Edem macular diabetic
Siguranța și eficacitatea clinică a afliberceptului au fost evaluate în două studii clinice randomizate, multicentrice, dublu-orb, controlate activ, la pacienți cu EMD (VIVIDDME și VISTADME). Un total de 862 pacienți au fost tratați și evaluați din punct de vedere al eficacității, 576 cu aflibercept.
Vârsta pacienților a variat între 23 ani și 87 ani, cu o medie de 63 ani. În studiile clinice efectuate cu pacienți cu EMD, aproximativ 47% (268/576) dintre pacienții randomizați la tratamentul cu aflibercept aveau vârsta de 65 ani sau mai mult și aproximativ 9% (52/576) aveau vârsta de 75 ani sau mai mult. Majoritatea pacienților din ambele studii clinice aveau diabet zaharat de tip II.
În ambele studii clinice, pacienții au fost repartizați randomizat, în raport de 1:1:1, pentru unul din cele 3 regimuri de dozare:
- Aflibercept administrat în doză de 2 mg la intervale de 8 săptămâni, după administrarea unei injecții lunare, timp de 5 luni consecutiv (aflibercept 2Q8);
- Aflibercept administrat în doză de 2 mg la intervale de 4 săptămâni (aflibercept 2Q4);
- Foto-coagulare cu laser la nivel macular (control cu tratament activ).
Începând cu săptămâna 24, pacienții care îndeplineau condiția pentru un prag prespecificat de pierdere a vederii erau eligibili pentru a li se administra tratament suplimentar: pacienții din grupurile tratate cu aflibercept puteau fi tratați cu laser, iar pacienții din grupul de control puteau fi tratați cu aflibercept.
În ambele studii clinice, obiectivul principal al eficacității a fost modificarea medie a AVOC de la momentul inițial până în săptămâna 52, atât grupul tratat cu aflibercept 2Q8, cât și grupul tratat cu aflibercept 2Q4 au demonstrat eficacitate superioară statistic față de grupul de control. Acest beneficiu s-a menținut până în săptămâna 100.
Rezultatele detaliate provenind din analiza studiilor clinice VIVIDDME și VISTADME sunt prezentate în Tabelul 5 și Figura 4 de mai jos.
Tabelul 5: Rezultatele privind eficacitatea în săptămâna 52 și săptămâna 100 (Set complet de analiză cu LOCF) în studiile clinice VIVIDDME și
VISTADME
| Rezultate privind eficacitatea | Studiul VIVIDDME | Studiul VISTADME | ||||||||||
| 52 săptămâni | 100 săptămâni | 52 săptămâni | 100 săptămâni | |||||||||
| Aflibercept 2 mg Q8 A (N = 135) | Aflibercept 2 mg Q4 (N = 136) | Control activ (laser) (N = 132) | Aflibercep t 2 mg Q8 A (N = 135) | Aflibercept 2 mg Q4 (N = 136) | Control activ (laser) (N = 132) | Aflibercept 2 mg Q8 A (N = 151) | Aflibercept 2 mg Q4 (N = 154) | Control activ (laser) (N = 154) | Aflibercept 2 mg Q8 A (N=151) | Aflibercep t 2 mg Q4 (N=154) | Control activ (laser) (N=154) | |
| Modificarea medie a AVOC măsurată prin scorul literelor ETDRS E față de momentul inițial | 10,7 | 10,5 | 1,2 | 9,4 | 11,4 | 0,7 | 10,7 | 12,5 | 0,2 | 11,1 | 11,5 | 0,9 |
| Diferența valorii medii a LSB,C,E (IÎ 97,5%) | 9,1 (6,4; 11,8) | 9,3 (6,5; 12,0) | 8,2 (5,2; 11,3) | 10,7 (7,6; 13,8) | 10,45 (7,7; 13,2) | 12,19 (9,4; 15,0) | 10,1 (7,0; 13,3) | 10,6 (7,1; 14,2) | ||||
| Procentul pacienților care au câștigat ≥ 15 litere din momentul inițial | 33% | 32% | 9% | 31,1% | 38,2% | 12,1% | 31% | 42% | 8% | 33,1% | 38,3% | 13,0% |
| Diferența ajustată D,C,E (IÎ 97,5%) | 24% (13,5; 34,9) | 23% (12,6; 33,9) | 19,0% (8,0; 29,9) | 26,1% (14,8; 37,5) | 23% (13,5; 33,1) | 34% (24,1; 44,4) | 20,1% (9,6; 30,6) | 25,8% (15,1; 36,6) | ||||
A După inițierea tratamentului cu 1 injecție administrată lunar, timp de 5 luni consecutiv
B Media LS și IÎ pe baza modelului ANCOVA cu determinarea AVOC la momentul inițial drept covariabilă și un factor pentru grupul de tratament. În plus, regiunea (Europa/Australia față de Japonia) a fost inclusă ca factor pentru studiul clinic VIVIDDME, iar antecedentele de IM și/sau AVC au fost incluse ca factor pentru studiul clinic VISTADME.
C Diferența este reprezentată de grupul tratat cu aflibercept minus grupul cu tratament de control activ (laser) D Diferența cu interval de încredere (IÎ) și testul statistic sunt calculate utilizând schema de ponderare Mantel-Haenszel ajustată în funcție de regiune (Europa/Australia față de Japonia) pentru studiul clinic VIVIDDME și antecedentele medicale de IM sau AVC pentru studiul clinic VISTADME E AVOC: Acuitatea vizuală optim corectată
ETDRS: Early Treatment Diabetic Retinopathy Study (Studiul retinopatiei diabetice cu tratament precoce) LOCF: Extrapolarea în sens longitudinal a ultimelor date observate
LS: Media celor mai mici pătrate derivată din ANCOVA IÎ: Interval de încredere
52
Figura 4: Modificarea medie a AVOC măsurată prin scorul literelor ETDRS de la momentul inițial până în săptămâna 100 în studiile clinice VIVIDDME și VISTADME
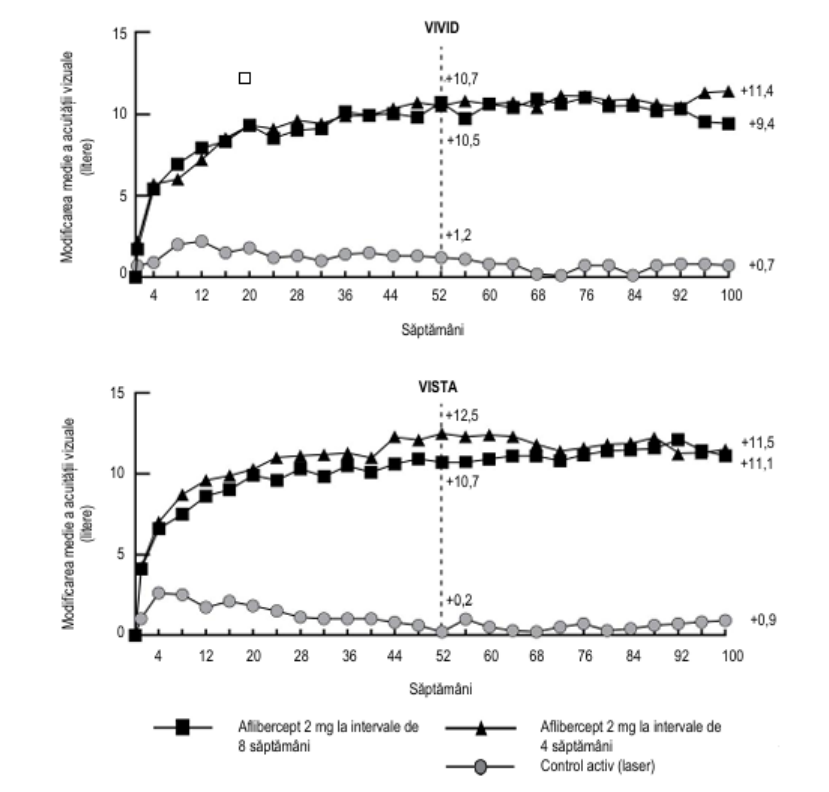
Efectele tratamentului în toate subgrupele evaluabile (de exemplu vârstă, sex, rasă, valoare HbA1c la momentul inițial, acuitate vizuală la momentul inițial, tratament anti-VEGF anterior) în fiecare studiu și în analiza combinată au fost, în general, în concordanță cu rezultatele obținute la populațiile globale.
În studiile clinice VIVIDDME și VISTADME, 36 pacienți (9%) și, respectiv, 197 pacienți (43%) au primit anterior tratament anti-VEGF, cu o perioadă de eliminare cu durata de 3 luni sau mai lungă. Efectele tratamentului la subgrupul de pacienți tratați cu un inhibitor de VEGF au fost similare cu efectele observate la pacienții naivi, care nu fuseseră tratați cu un inhibitor de VEGF.
Pacienții cu patologie bilaterală au fost eligibili pentru a li se administra tratament anti-VEGF la celălalt ochi, dacă medicul a considerat că este necesar acest lucru. În studiul clinic VISTADME, la 217 (70,7%) dintre pacienții cu aflibercept li s-au administrat injecții bilaterale cu aflibercept până în săptămâna 100; în studiul clinic VIVIDDME, la 97 (35,8%) dintre pacienții tratați cu aflibercept li s-a administrat un tratament anti- VEGF diferit la celălalt ochi.
Un studiu clinic comparativ independent (DRCR.net Protocol T) a utilizat un regim de dozare flexibil pe baza unor criterii stricte ale TCO și a criteriilor de re-tratament privind acuitatea vizuală. În grupul care a primit tratament cu aflibercept (n = 224), în săptămâna 52, acest regim a condus la o medie de 9,2 doze administrate la acești pacienți, similar cu numărul de doze administrate în grupul tratat cu aflibercept 2Q8 în studiile clinice VIVIDDME și VISTADME, în timp ce eficacitatea totală a grupului care a primit tratament cu aflibercept în studiul clinic Protocol T a fost comparabilă cu cea a grupului care a primit tratament cu aflibercept 2Q8 în studiile clinice VIVIDDME și VISTADME. În studiul clinic Protocolul T a fost observată o creștere medie de 13,3 litere a acuității vizuale, 42% dintre pacienți câștigând cel puțin 15 litere în acuitatea vizuală față de momentul inițial. Rezultatele privind siguranța au demonstrat că incidența generală a evenimentelor adverse oculare și non-oculare (inclusiv EAT) au fost comparabile în toate grupurile de tratament ale studiilor și între studii.
VIOLET, un studiu clinic de 100 de săptămâni, multicentric, randomizat, în regim deschis, controlat cu comparator activ la pacienți cu EMD a comparat trei regimuri diferite de dozare a aflibercept 2 mg pentru tratamentul EMD după cel puțin un an de tratament la intervale fixe, când tratamentul a fost inițiat cu 5 doze lunare consecutive, urmate de doze la fiecare 2 luni. Studiul a evaluat non- inferioritatea aflibercept 2 mg dozat conform regimului “tratament și extindere” (2T&E când intervalele de injectare au fost ținute la un minimum de 8 săptămâni și extins gradual pe baza rezultatelor clinice și anatomice) și aflibercept 2 mg dozat la nevoie (2PRN când pacienții au fost observați la fiecare 4 săptămâni și le-au fost administrate injecții la nevoie pe baza rezultatelor clinice și anatomice), comparat cu aflibercept 2 mg dozat la fiecare 8 săptămâni (2Q8) pentru al doilea și al treilea an de tratament.
Obiectivul primar de evaluare a eficacității (modificarea AVOC față de nivelul inițial în săptămâna 52) a fost 0,5 ± 6,7 litere în grupul 2T&E și 1,7 ± 6,8 litere în grupul 2PRN comparativ cu 0,4 ± 6,7 litere în grupul 2Q8, obținând non-inferioritate statistică (p<0,0001 pentru ambele comparații; limita NI 4 litere). Modificările AVOC față de nivelul de bază la săptămâna 100 erau consistente cu rezultatele săptămânii 52: - 0,1 ± 9,1 litere în grupul 2T&E și 1.8 ± 9.0 în grupul 2PRN comparat cu 0.1 ±
7.2 litere în grupul 2Q8. Media numărului de injectări peste 100 săptămâni a fost de 12,3, 10,0 și 11,5 pentru 2Q8fix, 2T&E și respectiv 2PRN.
Profilurile de siguranță oculară și sistemică în toate cele 3 grupuri de tratament au fost similare cu cele observate în studiile pivot VIVID și VISTA.
În grupul 2T&E, incrementele și decrementele pentru intervalele de injectare au fost la discreția investigatorului; ajustările de 2 săptămâni au fost recomandate în cadrul studiului
Neovascularizație coroidală miopică
Siguranța și eficacitatea aflibercept au fost evaluate în cadrul unui studiu randomizat, multicentric, cu dublă mascare a formei farmaceutice, controlat cu tratament fictiv, efectuat la pacienți asiatici cu NVC miopică, netratați anterior. Un total de 121 de pacienți au fost tratați și au fost evaluați din punct de vedere al eficacității (90 cu aflibercept). Vârsta pacienților a variat între 27 ani și 83 ani, cu o medie de 58 ani. În cadrul studiului efectuat la pacienți cu NVC miopică, aproximativ 36% (33/91) dintre pacienții randomizați la tratamentul cu aflibercept aveau vârsta de 65 ani sau mai mult, și aproximativ 10% (9/91) aveau vârsta de 75 ani sau mai mult.
Pacienții au fost repartizați aleator într-un raport de 3:1 pentru a primi fie 2 mg aflibercept intravitrean, fie injecții cu tratament fictiv, administrate la începutul studiului, injecții suplimentare administrându-se lunar în cazul persistenței sau recurenței bolii, până la săptămâna 24, când s-a evaluat obiectivul primar. La săptămâna 24, pacienții randomizați inițial pentru tratamentul fictiv au fost eligibili pentru a primi prima doză de aflibercept. În urma acesteia, pacienții din ambele grupuri au fost eligibili în continuare pentru injecții suplimentare în cazul persistenței sau recurenței bolii.
Diferența dintre grupurile de tratament a fost semnificativă statistic în favoarea aflibercept pentru obiectivul primar (modificarea AVOC) și obiectivul secundar de confirmare privind eficacitatea (proporția de pacienți la care s-a înregistrat un câștig de 15 litere din AVOC) la săptămâna 24 comparativ cu momentul inițial. Diferențele pentru ambele obiective s-au menținut până la săptămâna 48.
Rezultatele detaliate ale analizei din cadrul studiului clinic MYRROR sunt prezentate în Tabelul 6 și Figura 5 de mai jos.
Tabelul 6: Rezultatele privind eficacitatea în săptămâna 24 (analiza primară) și săptămâna 48 în studiul clinic MYRROR (Set complet de analiză cu LOCFA))
| Rezultate privind eficacitatea | Studiul MYRROR | |||
| 24 săptămâni | 48 săptămâni | |||
| Aflibercept 2 mg (N = 90) | Tratament fictiv (N = 31) | Aflibercept 2 mg (N = 90) | Tratament fictiv/ Aflibercept 2 mg (N = 31) | |
| Modificarea medie a AVOCB)măsurată prin scorul literelor ETDRS față de momentul inițial (DS) E) | 12,1 (8,3) | -2,0 (9,7) | 13,5 (8,8) | 3,9 (14,3) |
| Diferența în valoarea medie a LSC,D,E) (IÎ 95%) | 14,1 (10,8; 17,4) | 9,5 (5,4; 13,7) | ||
| Proporția de pacienți cu un câștig ≥15 litere de la momentul inițial | 38,9% | 9,7% | 50,0% | 29,0% |
| Diferența ponderată D,F) (IÎ 95%) | 29,2% (14,4; 44,0) | 21,0% (1,9; 40,1) | ||
A) LOCF: Extrapolarea în sens longitudinal a ultimelor date observate
B) AVOC: Acuitatea vizuală optim corectată
ETDRS: Early Treatment Diabetic Retinopathy Study (Studiul retinopatiei diabetice cu tratament precoce) DS: Deviație standard
C) Media LS: Media celor mai mici pătrate derivată din ANCOVA
D) IÎ:Interval de încredere
E) Diferența în valoarea medie LS și IÎ 95% pe baza unui model ANCOVA cu factori de tipul grup de tratament și țară (indicativele țărilor) și valoarea AVOC de la momentul inițial drept covariabilă.
F) Diferența și IÎ 95% CI sunt calculate utilizând testul Cochran-Mantel-Haenszel (CMH) ajustat pentru țară (indicativele țărilor)
Figura 5: Modificarea Medie a Acuității Vizuale în Funcție de Grupul de Tratament, de la Momentul Inițial până în Săptămâna 48, în Studiul Clinic MYRROR
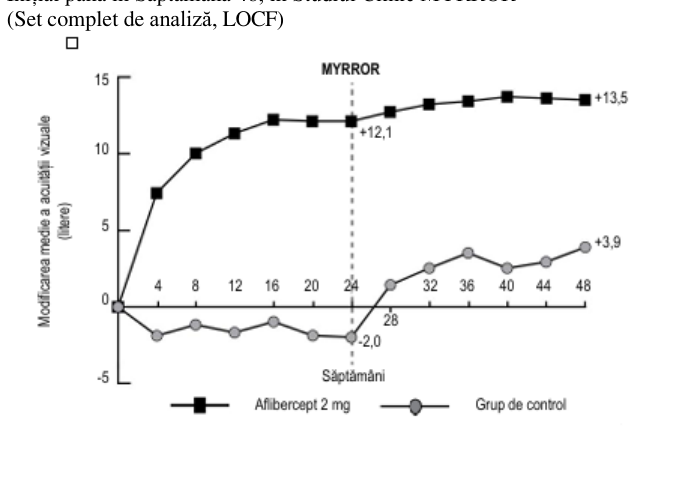
5.2 Proprietăți farmacocinetice
MYNZEPLI se administrează direct în corpul vitros, pentru a exercita efecte locale asupra ochiului.
Absorbție/distribuție
După administrarea intravitreană, aflibercept este absorbit lent de la nivelul ochiului în circulația sistemică și se observă predominant în circulația sistemică sub forma unui complex inactiv, stabil cu VEGF; cu toate acestea, numai „aflibercept liber” se poate lega de VEGF endogen.
Într-un sub-studiu farmacocinetic efectuat la 6 pacienți cu DMLV forma neovasculară (umedă) la care s-au recoltat frecvent probe, concentrațiile plasmatice maxime ale aflibercept liber (Cmax sistemică) au fost foarte scăzute, cu o medie de aproximativ 0,02 micrograme/ml (cuprinsă între 0 și 0,054) în decurs de 1 - 3 zile după injectarea intravitreană a dozei de 2 mg și nu au mai fost detectabile la două săptămâni după administrarea dozei, la aproape toți pacienții. Aflibercept nu se acumulează în plasmă când este administrat intravitrean la interval de 4 săptămâni.
Concentrația plasmatică maximă a aflibercept liber este de aproximativ 50 - 500 de ori mai mică decât concentrația de aflibercept necesară pentru inhibarea activității biologice a VEGF sistemic cu 50%, la modele animale la care s-au observat modificări ale tensiunii arteriale, după ce valorile circulante ale aflibercept liber au fost de aproximativ 10 micrograme/ml și au revenit la valorile inițiale, după ce valorile circulante ale aflibercept liber au scăzut sub aproximativ 1 microgram/ml. Se estimează că după administrarea intravitreană a dozei de 2 mg la pacienți, concentrația plasmatică maximă medie a aflibercept liber este de peste 100 de ori mai mică decât concentrația aflibercept necesară pentru legarea maximă a 50% din VEGF sistemic (2,91 micrograme/ml) într- un studiu efectuat cu voluntari sănătoși. Prin urmare, efectele farmacodinamice sistemice, cum sunt modificările tensiunii arteriale, sunt improbabile.
În cadrul unor sub-studii farmacocinetice la pacienți cu OVCR, ORVR, EMD sau NVC miopică valoarea medie a Cmax de aflibercept liber în plasmă a fost similară cu valorile din intervalul de 0,03 – 0,05 micrograme/ml și intervalul individual nu a depășit 0,14 micrograme/ml. După aceea, concentrațiile plasmatice pentru aflibercept liber au scăzut de la valori mai mici sau aproape de limita inferioară de cuantificare, în general, într-o săptămână; după 4 săptămâni, înainte de administrarea următoare, au fost atinse concentrații nedetectabile la toți pacienții.
Eliminare
Având în vedere faptul că MYNZEPLI este un medicament pe bază de proteine, nu s-au efectuat studii privind metabolizarea.
Aflibercept liber se leagă de VEGF pentru a forma un complex inert, stabil. Similar altor proteine cu molecule mari, se anticipează că atât aflibercept liber cât și cel legat vor fi eliminate prin catabolism proteolitic.
Insuficiență renală
Nu s-au efectuat studii specifice cu aflibercept la pacienți cu insuficiență renală.
Analiza farmacocinetică la pacienții incluși în studiul clinic VIEW2, dintre care 40% aveau insuficiență renală (24% ușoară, 15% moderată și 1% severă), nu a sugerat nicio diferență în ceea ce privește concentrațiile plasmatice ale medicamentului activ după administrarea intravitreană la intervale de 4 sau 8 săptămâni.
Rezultate similare au fost observate la pacienții cu OVCR în cadrul studiului clinic GALILEO, la pacienții cu EMD în cadrul studiului clinic VIVIDDME și la pacienții cu NVC miopică în studiul clinic MYRROR.
5.3 Date preclinice de siguranță
În studiile preclinice au fost observate efecte de toxicitate la doze repetate numai la expuneri sistemice considerate substanțial mai mari față de expunerea maximă la om după administrarea intravitreană în dozele clinice stabilite, fapt ce indică o relevanță clinică scăzută.
La maimuțele, cărora li s-a administrat aflibercept intravitrean, au fost observate eroziuni și ulcerații ale epiteliului respirator al cornetelor nazale la expuneri sistemice mai mari față de expunerea maximă la om. Expunerea sistemică bazată pe Cmax și ASC pentru aflibercept liber au fost de aproximativ 200 și, respectiv, de 700 de ori mai mari comparativ cu valorile corespunzătoare observate la om după administrarea intravitreană a unei doze de 2 mg. La o valoare a concentrației la care nu se observă nicio reacție adversă (NOAEL – No Observed Adverse Effect Level) de 0,5 mg/ochi la maimuțe, expunerea sistemică a fost de 42 și de 56 de ori mai mare, pe baza Cmax și, respectiv, a ASC.
Nu s-au efectuat studii privind potențialul mutagen sau carcinogen al aflibercept.
În cadrul studiilor privind dezvoltarea embriofetală la femele gestante de iepure, s-a demonstrat un efect al aflibercept asupra dezvoltării intrauterine în cazul administrării intravenoase (3 - 60 mg/kg) și subcutanate (0,1 mg/kg – 1 mg/kg). Valoarea la care nu se observă reacții adverse (NOAEL) materne a apărut la o doză de 3 mg/kg și, respectiv, de 1 mg/kg. Nu a fost identificat NOAEL legat de dezvoltare. La doza de 0,1 mg/kg, expunerea sistemică pe baza Cmax și ASC cumulativă pentru aflibercept liber a fost de aproximativ 17 și, respectiv, de 10 ori mai mare, comparativ cu valorile corespunzătoare observate la om după administrarea intravitreană a unei doze de 2 mg.
Efectele asupra fertilității masculine și feminine au fost evaluate în cadrul unui studiu cu durata de 6 luni, efectuat la maimuțe la care s-a administrat intravenos aflibercept în doze cuprinse între 3 și 30 mg/kg. La toate dozele s-au observat menstruații absente sau neregulate asociate cu modificări ale concentrațiilor hormonilor sexuali la femele și modificări ale morfologiei și motilității spermatozoizilor. Pe baza Cmax și ASC pentru aflibercept liber observate la administrarea de doze intravenoase de 3 mg/kg, expunerile sistemice au fost de aproximativ 4900 și, respectiv, de 1500 de ori mai mari comparativ cu valorile corespunzătoare observate la om după administrarea intravitreană a unei doze de 2 mg. Toate modificările au fost reversibile.
6. PROPRIETĂȚI FARMACEUTICE
6.1 Lista excipienților
L-histidină
Clorhidrat de L-histidină monohidrat
Trehaloză dihidrat
Poloxamer 188
Apă pentru preparate injectabile
6.2 Incompatibilități
În absența studiilor de compatibilitate, acest medicament nu trebuie amestecat cu alte medicamente.
6.3 Perioada de valabilitate
3 ani
6.4 Precauții speciale pentru păstrare
A se păstra la frigider (2°C - 8°C). A nu se congela.
A se păstra în ambalajul original pentru a fi protejat de lumină.
Flaconul nedeschis poate fi păstrat în afara frigiderului, sub 25°C timp de cel mult 24 ore. După deschiderea flaconului se continuă în condiții aseptice.
6.5 Natura și conținutul ambalajului
Soluție în flacon (sticlă de tip I) cu un dop (din cauciuc elastomeric bromobutilic) și un ac cu filtru de 18 G. Fiecare flacon conține un volum ce poate fi extras de cel puțin 0,1 ml. Mărimea ambalajului este de 1 flacon + 1 ac cu filtru.
6.6 Precauții speciale pentru eliminarea reziduurilor și alte instrucțiuni de manipulare
Flaconul este pentru utilizare unică, într-un singur ochi.
Flaconul conține mai mult decât doza recomandată de aflibercept 2 mg (echivalent cu 0,05 ml). Volumul în exces trebuie eliminat înainte de administrare.
Înainte de administrare, soluția trebuie inspectată vizual pentru a observa particulele străine și/sau modificările de culoare sau orice variație a aspectului fizic. În cazul în care se observă particule sau dacă soluția este tulbure sau prezintă modificări de culoare, medicamentul se aruncă.
Ac cu filtru:
Acul cu filtru cu vârf bont Blunt (Fill) nu este destinat injectării în piele. Nu utilizați autoclavul pentru acul cu filtru cu vârf bont Blunt (Fill).
Acul cu filtru este apirogen. Nu îl utilizați dacă ambalajul individual este deteriorat.
Aruncați acul cu filtru cu vârf bont Blunt (Fill) utilizat în recipiente aprobate pentru colectarea obiectelor ascuțite.
Atenție: Reutilizarea acului cu filtru poate duce la infectare sau alte afecțiuni/răni.
Pentru injectarea intravitreană trebuie utilizat un ac pentru injectare de 30 G x ½ inch.
Instrucțiuni pentru utilizarea flaconului:
- Se scoate capacul fără filet din plastic și se dezinfectează partea externă a dopului din cauciuc al flaconului.
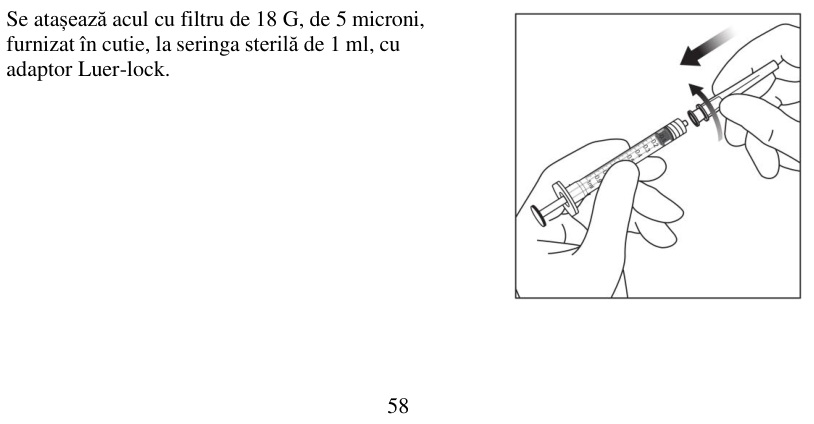
2.
- Se împinge acul cu filtru în centrul dopului flaconului până când acul este complet inserat în flacon și vârful atinge capătul inferior sau partea de jos a flaconului.
- Utilizând o tehnică aseptică, se extrage tot conținutul flaconului MYNZEPLI în seringă, menținând flaconul în poziție verticală și înclinându-l ușor pentru a facilita extragerea completă.
Pentru a împiedica introducerea de aer, asigurați-vă că suprafața oblică a acului cu filtru este scufundată în lichid. Continuați să înclinați flaconul pe parcursul extragerii menținând suprafața oblică a acului cu filtru scufundată în lichid.
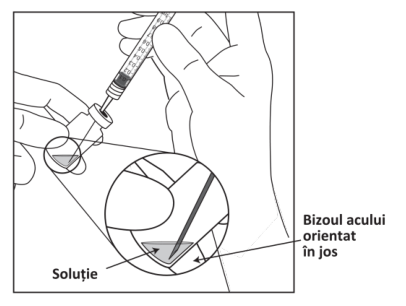
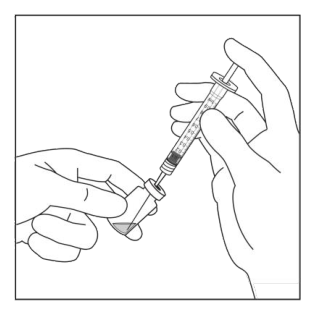
- Când se golește flaconul, se asigură că tija pistonului este retrasă suficient pentru a Permite golirea completă a acului cu filtru.
- Se scoate acul cu filtru și se elimină în mod adecvat.
Observație: Acul cu filtru nu trebuie utilizat pentru injecția intravitreană.
- Utilizând o tehnică aseptică, se înșurubează ferm acul de injecție 30 G x ½ inch la vârful seringii cu adaptor Luer-lock.
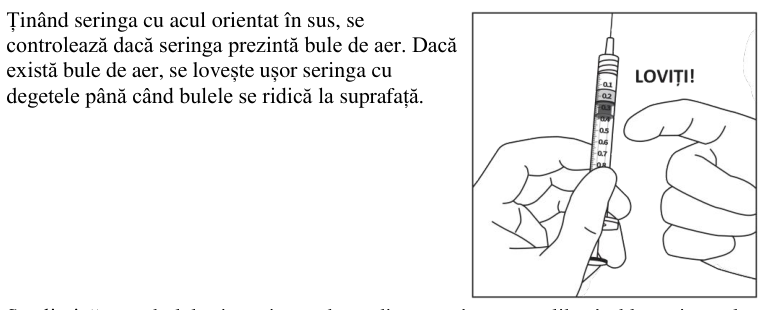
8.
- Se elimină toate bulele și cantitatea de medicament în exces, eliberând lent pistonul, astfel încât marginea plată a pistonului să se alinieze cu linia care marchează 0,05 ml pe seringă.
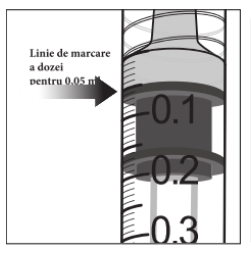
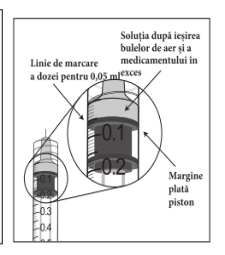
- Flaconul este numai pentru utilizare unică. Extragerea dozelor multiple dintr-un singur flacon poate crește riscul de contaminare și ulterior, infecție.
Orice medicament neutilizat sau material rezidual trebuie eliminat în conformitate cu reglementările locale.
7. DEȚINĂTORUL AUTORIZAȚIEI DE PUNERE PE PIAȚĂ
Advanz Pharma Limited
Unit 17 Northwood House
Northwood Crescent
Dublin 9
D09 V504
Irlanda
8. NUMĂRUL(ELE) AUTORIZAȚIEI DE PUNERE PE PIAȚĂ
EU/1/25/1964/001
9. DATA PRIMEI AUTORIZĂRI SAU A REÎNNOIRII AUTORIZAȚIEI
Data primei autorizări: 18 august 2025
10. DATA REVIZUIRII TEXTULUI
Informații detaliate privind acest medicament sunt disponibile pe site-ul Agenției Europene pentru Medicamente (EMA) https://www.ema.europa.eu.