IMDYLLTRA 10 mg
Rezumatul caracteristicilor produsului (RCP)
- 1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI
- 2. COMPOZIȚIA CALITATIVĂ ȘI CANTITATIVĂ
- 3. FORMA FARMACEUTICĂ
-
4. DATE CLINICE
- 4.1 Indicații terapeutice
- 4.2 Doze și mod de administrare
- 4.3 Contraindicații
- 4.4 Atenționări și precauții speciale pentru utilizare
- 4.5 Interacțiuni cu alte medicamente și alte forme de interacțiune
- 4.6 Fertilitatea, sarcina și alăptarea
- 4.7 Efecte asupra capacității de a conduce vehicule și de a folosi utilaje
- 4.8 Reacții adverse
- 4.9 Supradozaj
- 5. PROPRIETĂȚI FARMACOLOGICE
- 6. PROPRIETĂȚI FARMACEUTICE
- 7. DEȚINĂTORUL AUTORIZAȚIEI DE PUNERE PE PIAȚĂ
- 8. NUMĂRUL(ELE) AUTORIZAȚIEI DE PUNERE PE PIAȚĂ
- 9. DATA PRIMEI AUTORIZĂRI SAU A REÎNNOIRII AUTORIZAȚIEI
- 10. DATA REVIZUIRII TEXTULUI
1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI
IMDYLLTRA 1 mg pulbere pentru concentrat și soluție pentru soluție perfuzabilă
IMDYLLTRA 10 mg pulbere pentru concentrat și soluție pentru soluție perfuzabilă
2. COMPOZIȚIA CALITATIVĂ ȘI CANTITATIVĂ
IMDYLLTRA 1 mg pulbere pentru concentrat și soluție pentru soluție perfuzabilă
Un flacon de pulbere conține 1 mg de tarlatamab.
Reconstituirea cu apă pentru preparate injectabile are ca rezultat o concentrație finală de tarlatamab de 0,9 mg/ml.
IMDYLLTRA 10 mg pulbere pentru concentrat și soluție pentru soluție perfuzabilă
Un flacon de pulbere conține 10 mg de tarlatamab.
Reconstituirea cu apă pentru preparate injectabile are ca rezultat o concentrație finală de tarlatamab de 2,4 mg/ml.
Tarlatamab este produs în celule ovariene de hamster chinezesc prin tehnologia ADN-ului recombinant.
Excipient cu efect cunoscut
IMDYLLTRA conține 0,04 mg polisorbat 80 în fiecare flacon de 1 mg și 0,2 mg polisorbat 80 în fiecare flacon de 10 mg.
Pentru lista tuturor excipienților, vezi pct. 6.1.
3. FORMA FARMACEUTICĂ
Pulbere pentru concentrat și soluție pentru soluție perfuzabilă.
Tarlatamab pulbere (pulbere pentru concentrat): pulbere de culoare albă până la ușor gălbuie.
Soluție (stabilizator): soluție limpede, incoloră până la ușor gălbuie, cu un pH de 7,0.
4. DATE CLINICE
4.1 Indicații terapeutice
IMDYLLTRA este indicat ca monoterapie pentru tratamentul pacienților adulți cu cancer pulmonar cu celule mici în stadiu extins (CPCM-SE) care necesită terapie sistemică după progresia bolii în timpul sau după tratamentul de primă linie cu chimioterapie pe bază de platină.
4.2 Doze și mod de administrare
Tratamentul cu IMDYLLTRA trebuie inițiat sub îndrumarea și supravegherea unor medici cu experiență în utilizarea terapiei anticancer. Trebuie administrat într-o unitate medicală adecvată. Vezi tabelul 2 pentru medicamentele concomitente recomandate.
Pacienții trebuie monitorizați de la începutul perfuziei, timp de 6 până la 8 ore, în ziua 1 și ziua 8. Monitorizarea suplimentară și monitorizarea perfuziilor ulterioare sunt la discreția medicului.
În ziua 1 și în ziua 8, pacienții trebuie instruiți ca de la începutul fiecărei perfuzii să rămână timp de 24 ore în apropierea unei unități medicale adecvate, însoțiți de o persoană care are grijă de pacient.
Înainte de externare, atât pacienții, cât și persoanele care au grijă de pacienți trebuie informați cu privire la semnele și simptomele sindromului de eliberare de citokine (SEC) și ale sindromului de neurotoxicitate asociat celulelor efectoare imunitare (SNCEI).
Doze
Schema terapeutică recomandată pentru IMDYLLTRA prevede o doză inițială de 1 mg în ziua 1 urmată de 10 mg în zilele 8 și 15 și apoi la interval de 2 săptămâni, așa cum se arată în tabelul 1.
Pacienții trebuie tratați până la progresia bolii sau până la apariția unei toxicități inacceptabile.
Tabelul 1. Schema terapeutică recomandată pentru IMDYLLTRA
| Doza de IMDYLLTRA | |
|---|---|
| Ziua 1 | 1 mg |
| Ziua 8 | 10 mg |
| Ziua 15 și ulterior la interval de 2 săptămâni | 10 mg |
Medicamente concomitente recomandate
Pentru a reduce riscul sindromului de eliberare de citokine (vezi pct. 4.4), medicamentele concomitente pentru administrarea IMDYLLTRA trebuie utilizate așa cum se prezintă în tabelul 2.
Tabelul 2. Medicamente administrate concomitent în ziua 1 și ziua 8
| Ziua tratamentului | Medicamente | Administrare |
|---|---|---|
| Ziua 1 și ziua 8 | Administrați intravenos 8 mg de dexametazonă (sau un echivalent) | Cu 1 oră înainte de perfuzia cu IMDYLLTRA |
Conform ghidurilor de îngrijire standard, se recomandă administrarea intravenoasă a 1 litru de soluție injectabilă de clorură de sodiu 9 mg/ml (0,9%) | Imediat după finalizarea perfuziei cu IMDYLLTRA |
Reluarea administrării IMDYLLTRA după întârzierea administrării dozei
Dacă administrarea unei doze de IMDYLLTRA este întârziată, tratamentul trebuie reluat în conformitate cu recomandările prezentate în tabelul 3, iar schema de administrare trebuie reluată în consecință. Medicamentele concomitente recomandate trebuie administrate conform indicațiilor din tabelul 2.
Tabelul 3. Recomandări pentru reluarea tratamentului cu IMDYLLTRA după întârzierea administrării unei doze
| Ultima doză administrată | Timp de la ultima doză administrată | Acțiunea |
|---|---|---|
| 1 mg în ziua 1 | 2 săptămâni sau mai puțin (≤ 14 zile) | Administrați IMDYLLTRA 10 mg, apoi reluați schema terapeutică planificată. |
| Mai mult de 2 săptămâni (> 14 zile) | Administrați IMDYLLTRA 1 mg. Dacă tratamentul este tolerat, creșteți doza la 10 mg după 1 săptămână. Apoi reluați schema terapeutică planificată. | |
| 10 mg în ziua 8 | 3 săptămâni sau mai puțin (≤ 21 zile) | Administrați IMDYLLTRA 10 mg, apoi reluați schema terapeutică planificată. |
| Mai mult de 3 săptămâni (> 21 zile) | Administrați IMDYLLTRA 1 mg. Dacă tratamentul este tolerat, creșteți doza la 10 mg după 1 săptămână. Apoi reluați schema terapeutică planificată. | |
| 10 mg în ziua 15 și ulterior la fiecare 2 săptămâni | 4 săptămâni sau mai puțin (≤ 28 zile) | Administrați IMDYLLTRA 10 mg, apoi reluați schema terapeutică planificată. |
| Mai mult de 4 săptămâni (> 28 zile) | Administrați IMDYLLTRA 1 mg. Dacă tratamentul este tolerat, creșteți doza la 10 mg după 1 săptămână. Apoi reluați schema terapeutică planificată. |
a Administrați medicamentele concomitente recomandate înainte și după perfuziile cu IMDYLLTRA în ziua 1 și ziua 8 și monitorizați pacienții în mod corespunzător (vezi tabelul 2).
Modificările dozei și abordarea terapeutică a reacțiilor adverse
Nu se recomandă reducerea dozei de IMDYLLTRA.
Vezi tabelul 4 pentru acțiunile recomandate pentru abordarea terapeutică a SEC, tabelul 5 pentru acțiunile recomandate pentru abordarea terapeutică a SNCEI și tabelul 6 pentru abordarea terapeutică a altor reacții adverse.
Sindromul de eliberare de citokine (SEC)
SEC trebuie diagnosticat pe baza tabloului clinic (vezi pct. 4.4). Pacienții trebuie evaluați pentru a se determina dacă există alte cauze de febră, hipoxie și hipotensiune arterială și tratați în consecință. Dacă se suspectează SEC, acesta trebuie abordat terapeutic conform recomandărilor din tabelul 4. Pacienții care prezintă SEC de gradul 2 sau mai mare (de exemplu, hipotensiune arterială care nu răspunde la administrarea de fluide sau hipoxie care necesită administrarea de oxigen suplimentar) trebuie monitorizați pentru semnele și simptomele de SEC – inclusiv febră, hipotensiune arterială și hipoxie – utilizând pulsoximetrie sau telemetrie cardiacă, după cum este indicat. În cazurile de SEC sever sau care pune viața în pericol, se recomandă terapia anti-IL-6, de exemplu administrarea de tocilizumab și internarea într-o secție de terapie intensivă (ATI) pentru terapie de susținere.
Tabelul 4. Recomandări pentru clasificarea și abordarea terapeutică a sindromului de eliberare de citokine și pentru modificarea dozeia
| Gradul SEC | Simptome definitorii | Modificarea dozei de IMDYLLTRA | Abordare terapeutică |
|---|---|---|---|
| Grad 1 | Simptomele necesită doar tratament simptomatic (de exemplu, febră ≥ 38 °C fără hipotensiune arterială sau hipoxie). |
|
|
| Grad 2 | Simptomele necesită și răspund la intervenție moderată.
|
|
La reluarea tratamentului, la următoarea doză planificată, pacienții trebuie monitorizați, la latitudinea medicului, într-o unitate medicală adecvatăb. |
| Grad 3 | Simptome severe, definite ca temperatură ≥ 38 °C cu:
|
IMDYLLTRA. | Pe lângă tratamentul pentru gradul 2:
La reluarea tratamentului, la următoarea doză planificată, pacienții trebuie monitorizați, la latitudinea medicului, într-o unitate medicală adecvatăb. |
| Grad 4 | Simptome care pun viața în pericol, definite ca temperatură ≥ 38 °C cu:
| Opriți definitiv administrarea de IMDYLLTRA. |
|
a SEC conform clasificării consensuale ASTCT (American Society for Transplantation and Cellular Therapy), 2019.
b Vezi tabelul 3 pentru recomandări privind reluarea administrării IMDYLLTRA după întârzierea administrării dozelor.
c Reduceți treptat steroizii, conform ghidurilor de îngrijire standard.
ATI = secție de terapie intensivă
Sindromul de neurotoxicitate asociat celulelor efectoare imunitare (SNCEI)
Pacienții trebuie monitorizați pentru semne și simptome de SNCEI. Trebuie excluse alte cauze ale simptomelor neurologice. Trebuie asigurată terapie intensivă pentru toxicitățile neurologice severe sau care pun viața în pericol. Dacă se suspectează SNCEI, acesta trebuie abordat terapeutic conform recomandărilor din tabelul 5.
Tabelul 5. Recomandări pentru clasificarea și abordarea terapeutică a sindromului de neurotoxicitate asociat celulelor efectoare imunitare și pentru modificarea dozeia
| Gradul SNCEIa | Simptome definitorii | Modificarea dozei de IMDYLLTRA | Abordare terapeutică |
|---|---|---|---|
| Grad 1 | Scor ICE 7-9b fără nivel de conștiență redus. |
IMDYLLTRA la următoarea doză programatăc. |
|
| Grad 2 | Scor ICE 3-6b și/sau somnolență ușoară, trezire la auzul vocii. |
IMDYLLTRA la următoarea doză programatăc. |
IMDYLLTRAc. |
| Grad 3 | Scor ICE 0-2b și/sau nivel redus de conștiență, trezire doar la stimuli tactili și/sau orice criză convulsivă clinică focală sau generalizată care se remite rapid Sau Crize non- convulsive detectate la EEG care se remit după intervenție și/sau edem focal sau local observat la neuroimagistică. |
|
IMDYLLTRAc. |
| Grad 4 | Scor ICE 0b (pacientul nu răspunde la stimuli și nu poate efectua ICE) și/sau stupor sau comă și/sau convulsii prelungite care pun viața în pericol (> 5 minute) sau convulsii clinice sau electrice repetitive fără revenire la starea neurologică de bază între episoade și/sau edem cerebral difuz la neuroimagistică, postură de decerebrare sau de decorticare sau edem papilar, paralizie de nerv cranian VI sau triadă Cushing. |
IMDYLLTRA. |
|
a SNCEI conform clasificării consensuale ASTCT (American Society for Transplantation and Cellular Therapy), 2019.
b Dacă pacientul răspunde la stimuli și este capabil să efectueze testul de evaluare a encefalopatiei asociate cu celulele efectoare imunitare (ICE), se vor evalua: orientarea (orientare în ceea ce privește anul, luna, orașul, spitalul = 4 puncte); numirea (numește 3 obiecte, de exemplu arătați spre ceas, pix, nasture = 3 puncte); respectă indicațiile (de exemplu când spuneți „arătați-mi 2 degete” sau „închideți ochii și scoateți limba” = 1 punct); scrierea (capacitatea de a scrie o propoziție standard = 1 punct) și atenția (numărare inversă de la 100 din zece în zece = 1 punct). Dacă pacientul nu răspunde la stimuli și nu poate efectua evaluarea ICE (SNCEI grad 4) = 0 puncte.
c Vezi tabelul 3 pentru recomandări privind reluarea administrării IMDYLLTRA după întârzierea administrării dozelor.
d Reduceți treptat steroizii, conform ghidurilor de îngrijire standard.
CT = tomografie computerizată; EEG = electroencefalogramă; ATI = secție de terapie intensivă;
IRM = imagistică prin rezonanță magnetică
Neutropenie și alte reacții adverse
Neutropenia și alte reacții adverse trebuie abordate terapeutic în conformitate cu tabelul 6.
Tabelul 6. Întreruperi recomandate ale tratamentului cu IMDYLLTRA pentru abordarea terapeutică a altor reacții adversea,b
| Reacții adverse | Severitatea | Modificarea dozeib |
|---|---|---|
| Neutropenie (vezi pct. 4.4) | Gradele 1 și 2 | Nu este necesară întreruperea tratamentului. |
| Grad 3 |
| |
| Grad 4 |
IMDYLLTRA. Luați în considerare utilizarea factorului de stimulare a coloniilor granulocitare (FSCG). | |
| Hepatotoxicitate (vezi pct. 4.4)c | Grad 3 Valori crescute ale ALT sau AST sau ale bilirubinei |
|
| Grad 4 Valori crescute ale ALT sau AST sau ale bilirubinei |
| |
| AST sau ALT > 3 × LSN, cu bilirubina totală > 2 × LSN dacă nu există cauze alternative |
| |
| Alte reacții adverse (vezi pct. 4.8) | Grad 3 sau 4 | Întrerupeți administrarea IMDYLLTRA până la revenirea la gradul ≤ 1 sau la starea inițială. Luați în considerare oprirea definitivă dacă reacția adversă nu se remite în decurs de 28 zile.
|
a Severitate conform criteriilor standard NCI CTCAE (National Cancer Institute Common Terminology Criteria for Adverse Events), versiunea 5.0.
b Vezi tabelul 3 pentru recomandări privind reluarea administrării IMDYLLTRA după întârzierea administrării dozelor.
c La pacienții cu valori crescute ale enzimelor hepatice la momentul inițial, pentru evaluarea hepatotoxicității trebuie utilizați multipli ai valorilor de la momentul inițial.
ALT = alanin-aminotransferază; AST = aspartat-aminotransferază; LSN = limita superioară a valorilor normale
Grupe speciale de pacienți
Vârstnici
Nu este necesară ajustarea dozei la pacienții vârstnici (vârsta ≥ 65 ani).
Insuficiență hepatică
Nu este necesară ajustarea dozei la pacienții cu insuficiență hepatică ușoară (vezi pct. 5.2). Sunt disponibile date limitate privind pacienții cu insuficiență hepatică moderată. IMDYLLTRA nu a fost studiat la pacienți cu insuficiență hepatică severă. Nu se pot face recomandări privind doza la pacienții cu insuficiență hepatică moderată sau severă.
Insuficiență renală
Nu este necesară ajustarea dozei la pacienții cu insuficiență renală ușoară sau moderată (vezi pct. 5.2). IMDYLLTRA nu a fost studiat la pacienți cu insuficiență renală severă. Nu se pot face recomandări privind doza la pacienții cu insuficiență renală severă până la boală renală în stadiu terminal.
Copii și adolescenți
IMDYLLTRA nu prezintă utilizare relevantă la copii și adolescenți pentru tratamentul cancerului pulmonar cu celule mici.
Mod de administrare
IMDYLLTRA este destinat administrării intravenoase.
IMDYLLTRA trebuie reconstituit și apoi diluat suplimentar, înainte de administrarea prin perfuzie intravenoasă.
Pentru instrucțiuni privind reconstituirea și diluarea medicamentului înainte de administrare, vezi pct. 6.6.
Linia de perfuzie pentru premedicație poate fi utilizată pentru IMDYLLTRA. Trebuie efectuată o spălare a liniei de perfuzie, între administrarea medicamentelor concomitente și administrarea IMDYLLTRA.
Administrați întregul conținut de IMDYLLTRA ca perfuzie intravenoasă cu durata de 1 oră, cu debit constant, utilizând o pompă de perfuzie; vezi tabelul 7. Pompa trebuie să fie programabilă, blocabilă, neelastomerică și să aibă o alarmă.
Linia de perfuzie este amorsată cu soluție injectabilă de clorură de sodiu 9 mg/ml (0,9%) SAU cu preparatul final IMDYLLTRA.
După finalizarea perfuziei cu IMDYLLTRA, linia de perfuzie intravenoasă trebuie spălată timp de 3-5 minute utilizând soluție injectabilă de clorură de sodiu 9 mg/ml (0,9%).
Tabelul 7. Informații privind administrarea tarlatamab
| Durata de perfuzare a 250 ml de preparat intravenos | Debit de perfuzare (ml/oră) |
|---|---|
| 1 oră | 250 ml/oră |
4.3 Contraindicații
Hipersensibilitate la substanța activă sau la oricare dintre excipienții enumerați la pct. 6.1.
4.4 Atenționări și precauții speciale pentru utilizare
Trasabilitate
Pentru a avea sub control trasabilitatea medicamentelor biologice, numele și numărul lotului medicamentului administrat trebuie înregistrate cu atenție.
Sindromul de eliberare de citokine (SEC)
Administrarea de tarlatamab a fost asociată cu SEC, inclusiv cu evenimente care pot pune viața în pericol sau pot fi letale; vezi pct. 4.8. SEC poate fi asociat cu simptome precum febră, hipotensiune arterială, hipoxie, oboseală, tahicardie, cefalee, frisoane, greață și vărsături.
Pacienții și persoanele care au grijă de pacienți trebuie informați cu privire la posibilitatea apariției SEC după externare și instruiți să solicite imediat asistență medicală dacă apar semne sau simptome.
Tarlatamab trebuie administrat într-o unitate medicală dotată cu echipament pentru monitorizarea și abordarea terapeutică a SEC. Înainte de începerea perfuziilor trebuie să vă asigurați că pacienții sunt normovolemici. Pacienții trebuie monitorizați atent pentru semne și simptome de SEC în timpul inițierii tratamentului cu tarlatamab. Pentru a atenua riscul de SEC, este important ca tratamentul cu tarlatamab să se înceapă cu doza inițială recomandată în tabelul 1.
SEC trebuie abordat terapeutic conform recomandărilor din tabelul 4.
Sindromul de neurotoxicitate asociat celulelor efectoare imunitare (SNCEI)
Administrarea de tarlatamab a fost asociată cu SNCEI, inclusiv cu evenimente care pot pune viața în pericol sau pot fi letale; vezi pct. 4.8. SNCEI poate apărea și după câteva săptămâni de la administrarea tarlatamab. Reacțiile adverse care pot fi asociate cu SNCEI includ cefalee, encefalopatie, confuzie, delir, convulsii, ataxie, neurotoxicitate și tremor. Pacienții trebuie monitorizați atent pentru semne și simptome de SNCEI în timpul tratamentului cu tarlatamab.
Pacienții și persoanele care au grijă de pacienți trebuie informați cu privire la posibilitatea apariției SNCEI după externare și instruiți să solicite imediat asistență medicală dacă apar semne sau simptome.
SNCEI trebuie abordat terapeutic conform recomandărilor din tabelul 5.
Neutropenie
Administrarea de tarlatamab a fost asociată cu neutropenie; vezi pct. 4.8. Pacienții trebuie monitorizați atent pentru semne și simptome de neutropenie în timpul tratamentului cu tarlatamab.
Neutropenia trebuie abordată terapeutic conform recomandărilor din tabelul 6.
Infecții
Infecții grave, inclusiv infecții care pun viața în pericol și infecții letale, au fost raportate la pacienții tratați cu tarlatamab. Cele mai frecvente infecții includ pneumonia, infecția tractului urinar, COVID-19, infecția tractului respirator superior, infecția tractului respirator, infecția cu candida, candidoza orală și rinofaringita.
Pacienții trebuie monitorizați pentru semne și simptome de infecții înaintea și în timpul tratamentului cu tarlatamab.
Hipersensibilitate
La pacienții tratați cu tarlatamab au fost raportate reacții de hipersensibilitate, inclusiv evenimente severe rare. Semnele și simptomele clinice de hipersensibilitate pot include – dar nu se limitează la – erupție cutanată tranzitorie și bronhospasm. În timpul tratamentului cu tarlatamab, pacienții trebuie monitorizați pentru semne și simptome de hipersensibilitate și tratați conform indicațiilor clinice. Trebuie luate în considerare întreruperea sau oprirea definitivă a tratamentului cu tarlatamab, în funcție de severitate; vezi tabelul 6 pentru abordarea terapeutică a altor reacții adverse.
Hepatotoxicitate
Administrarea de tarlatamab a fost asociată cu valori crescute ale enzimelor hepatice. O valoare crescută a enzimelor hepatice poate să apară cu sau fără prezența simultană a SEC.
Valorile enzimelor hepatice și ale bilirubinei trebuie monitorizate înainte de tratamentul cu tarlatamab și conform indicațiilor clinice. Posibilele toxicități trebuie abordate terapeutic conform recomandărilor din tabelul 6.
Femei aflate la vârsta fertilă/contracepție
Prezența unei sarcini la femeile aflate la vârsta fertilă trebuie verificată înainte de a începe tratamentul cu tarlatamab. Femeile aflate la vârsta fertilă trebuie să utilizeze măsuri contraceptive eficace în timpul tratamentului și timp de 2 luni după ultima doză de tarlatamab (vezi pct. 4.6).
Excipienți cu efect cunoscut
Acest medicament conține sodiu mai puțin de 1 mmol (23 mg) per doză, adică practic „nu conține sodiu”.
Acest medicament conține 0,04 mg de polisorbat 80 în fiecare flacon de 1 mg și 0,2 mg în fiecare flacon de 10 mg. Polisorbații pot provoca reacții alergice.
4.5 Interacțiuni cu alte medicamente și alte forme de interacțiune
Nu s-au efectuat studii privind interacțiunile. Inițierea tratamentului cu tarlatamab determină eliberarea tranzitorie de citokine, care pot inhiba enzimele CYP450 și pot duce la expuneri crescute la substraturile CYP administrate concomitent. Pacienții tratați concomitent cu substraturi CYP450, în special cele cu un indice terapeutic îngust, trebuie monitorizați pentru reacțiile adverse cunoscute. Doza medicamentului administrat concomitent trebuie ajustată, după cum este necesar.
4.6 Fertilitatea, sarcina și alăptarea
Femei aflate la vârsta fertilă/contracepție
Femeile aflate la vârsta fertilă trebuie să utilizeze măsuri contraceptive eficace în timpul tratamentului și timp de 2 luni după tratament cu tarlatamab.
Sarcina
Nu există date disponibile privind utilizarea tarlatamab la femeile gravide.
Un studiu cu privire la toxicitatea asupra funcției de reproducere efectuat la șoareci utilizând molecula surogat murină muS757 a arătat transportul transplacentar al muS757 (vezi pct. 5.3). Având în vedere mecanismul de acțiune al tarlatamabului și potențiala dezvoltare de reacții adverse (cum ar fi SEC) în urma expunerii la tarlatamab, acesta poate provoca leziuni fetale atunci când este administrat unei femei gravide (vezi pct. 5.1).
Tarlatamab nu este recomandat în timpul sarcinii și la femei aflate la vârsta fertilă care nu utilizează măsuri contraceptive.
Înainte de a începe tratamentul cu tarlatamab trebuie verificat dacă femeile aflate la vârsta fertilă sunt gravide.
Alăptarea
Nu se cunoaște dacă tarlatamab este secretat în laptele matern. Deoarece multe medicamente, inclusiv anticorpii, pot fi secretate în laptele matern, nu se poate exclude un risc pentru nou-născuți/sugari. Alăptarea trebuie întreruptă în timpul tratamentului cu tarlatamab și timp de cel puțin 2 luni după ultima doză.
Fertilitatea
Nu există studii clinice care să evalueze efectul tarlatamabului asupra fertilității.
4.7 Efecte asupra capacității de a conduce vehicule și de a folosi utilaje
Din cauza potențialului de evenimente neurologice asociate cu SNCEI ca urmare a perfuziei cu tarlatamab, tarlatamab poate avea o influență majoră asupra capacității de a conduce vehicule și de a folosi utilaje. În cazul apariției oricăror simptome neurologice, pacienții trebuie sfătuiți să se abțină de la a conduce vehicule și de la a se angaja în ocupații sau activități periculoase, cum ar fi folosirea de utilaje grele sau potențial periculoase, până la dispariția simptomelor.
4.8 Reacții adverse
Rezumatul profilului de siguranță
Siguranța IMDYLLTRA a fost evaluată în studii clinice la 473 pacienți cu cancer pulmonar cu celule mici (CPCM) cărora li s-a administrat doza țintă de tarlatamab de 10 mg în monoterapie.
Cele mai frecvente reacții adverse sunt: SEC (56,7%), scăderea poftei de mâncare (36,4%), febra (31,9%), disgeuzia (31,3%), constipația (30,4%), anemia (30,0%), oboseala (29,8%), greața (24,9%), astenia (19,0%), neutropenia (16,9%), hiponatremia (16,7%), cefaleea (16,3%), limfopenia (15,6%).
Cele mai frecvente reacții adverse grave sunt SEC (19,7%) și febra (4,7%).
Rezumatul reacțiilor adverse sub formă de tabel
Reacțiile adverse raportate în studiile clinice sunt enumerate în funcție de clasa de sisteme și organe și de frecvență. Frecvențele reacțiilor adverse se bazează pe date cumulate de la 473 pacienți dintr-un studiu clinic de fază 1, un studiu clinic de fază 2 și un studiu clinic de fază 3. Durata mediană a expunerii a fost de 18,0 săptămâni (interval: 0,1-175,1 săptămâni).
Reacțiile adverse sunt enumerate conform clasificării MedDRA, pe aparate, sisteme și organe și în funcție de frecvență. În funcție de categoriile de frecvență, ele sunt definite ca fiind foarte frecvente (≥ 1/10), frecvente (≥ 1/100 și < 1/10), mai puțin frecvente (≥ 1/1 000 și < 1/100), rare (≥ 1/10 000 și < 1/1 000), foarte rare (< 1/10 000) și cu frecvență necunoscută (frecvența nu poate fi estimată din datele disponibile). În cadrul fiecărei grupe de frecvență, reacțiile adverse sunt prezentate în ordinea descrescătoare a gravității.
Tabelul 8. Reacții adverse
| MedDRA pe aparate, sisteme și organe | Reacție adversă | Toate gradele | Grad ≥ 3 |
|---|---|---|---|
| Tulburări hematologice și limfatice | Anemie | Foarte frecvente | Frecvente |
| Neutropeniea, c | Foarte frecvente | Frecvente | |
| Limfocitopenieb | Foarte frecvente | Foarte frecvente | |
| Trombocitopenie | Frecvente | Mai puțin frecvente | |
| Leucopenie | Frecvente | Mai puțin frecvente | |
| Tulburări gastro-intestinale | Constipație | Foarte frecvente | Mai puțin frecvente |
| Greață | Foarte frecvente | Mai puțin frecvente | |
| Vărsături | Foarte frecvente | Mai puțin frecvente | |
| Diaree | Foarte frecvente | Mai puțin frecvente | |
| Tulburări generale și la nivelul locului de administrare | Pirexie | Foarte frecvente | Mai puțin frecvente |
| Oboseală | Foarte frecvente | Frecvente | |
| Astenie | Foarte frecvente | Frecvente | |
| Frisoane | Frecvente | Nu au fost raportate | |
| Tulburări ale sistemului imunitar | Sindromul de eliberare de citokinec | Foarte frecvente | Frecvente |
| Investigații diagnostice | Scădere ponderală | Foarte frecvente | Frecvente |
| Valoare crescută a alanin- aminotransferazei | Foarte frecvente | Frecvente | |
| Valoare crescută a aspartat- aminotransferazei | Frecvente | Frecvente | |
| Număr scăzut de leucocite | Frecvente | Frecvente | |
| Tulburări metabolice și de nutriție | Scădere a poftei de mâncare | Foarte frecvente | Frecvente |
| Hiponatremie | Foarte frecvente | Frecvente | |
| Hipokalemie | Foarte frecvente | Frecvente | |
| Hipomagneziemie | Frecvente | Mai puțin frecvente | |
| Tulburări musculo-scheletice și ale țesutului conjunctiv | Mialgie | Frecvente | Nu au fost raportate |
| Tulburări ale sistemului nervos | Disgeuzie | Foarte frecvente | Nu au fost raportate |
| Cefalee | Foarte frecvente | Nu au fost raportate | |
| Amețeală | Frecvente | Nu au fost raportate | |
| Sindromul de neurotoxicitate asociat celulelor efectoare imunitarec | Frecvente | Mai puțin frecvente | |
| Tremor | Frecvente | Nu au fost raportate | |
| Neurotoxicitate | Mai puțin frecvente | Nu au fost raportate | |
| Convulsii | Mai puțin frecvente | Mai puțin frecvente | |
| Ataxie | Mai puțin frecvente | Mai puțin frecvente | |
| Encefalopatie | Mai puțin frecvente | Mai puțin frecvente | |
| Tulburări psihice | Stare de confuzie | Frecvente | Mai puțin frecvente |
| Delir | Frecvente | Mai puțin frecvente | |
| Tulburări respiratorii, toracice și mediastinale | Dispnee | Foarte frecvente | Frecvente |
| Afecțiuni cutanate și ale țesutului subcutanat | Prurit | Foarte frecvente | Mai puțin frecvente |
| Erupție cutanată tranzitorie | Frecvente | Mai puțin frecvente | |
| Tulburări vasculare | Hipotensiune arterială | Frecvente | Frecvente |
| Hipertensiune arterială | Frecvente | Frecvente |
a Include scăderea numărului de neutrofile.
b Include scăderea numărului de limfocite.
c Informații suplimentare sunt furnizate în „Descrierea reacțiilor adverse selectate”.
Descrierea reacțiilor adverse selectate
Sindromul de eliberare de citokine (SEC)
În studiile clinice cu date privind siguranța cumulate de la 473 pacienți cu CPCM la care s-a administrat IMDYLLTRA 1 mg ca primă doză și 10 mg ca a doua doză și doze ulterioare, SEC a apărut la 56,7% dintre pacienți, cu evenimente de gradul 1 la 39,3% dintre pacienți, de gradul 2 la 15,4% dintre pacienți, de gradul 3 la 1,7% dintre pacienți și de gradul 4 la 0,2% dintre pacienți. Evenimente grave de SEC au fost raportate la 19,7% dintre pacienți. După prima doză de IMDYLLTRA, 41,4% dintre pacienți au prezentat SEC de orice grad, iar 34,0% dintre pacienți au prezentat SEC de orice grad după a doua doză. Majoritatea evenimentelor SEC au apărut după primele două doze, 8,5% dintre pacienți prezentând SEC după a treia doză sau mai târziu. După perfuzia din ziua 1, 13,7% dintre pacienți au prezentat SEC de grad ≥ 2. După perfuzia din ziua 8, 4,4% dintre pacienți au prezentat SEC de grad ≥ 2. Timpul median de la cea mai recentă doză de IMDYLLTRA până la prima apariție a SEC a fost de 15,9 ore (interval: de la 9,0 până la 26,5 ore). Pentru evenimentele de gradul 1 care au progresat la gradul 2 sau mai mult, timpul median de la evenimentul de grad 1 la evenimentele de grad 2 sau mai mare a fost de 22,1 ore (interval intercuartilic: 8,5-31,6 ore). Sindromul de eliberare de citokine a dus la întreruperea tratamentului și/sau la modificarea dozei la 2,1% dintre pacienți și la oprirea tratamentului cu tarlatamab la 0,6% dintre pacienți.
Au fost raportate cazuri letale de SEC în contextul ulterior punerii pe piață.
Pentru abordarea terapeutică clinică a SEC, vezi pct. 4.4.
Sindromul de neurotoxicitate asociat celulelor efectoare imunitare (SNCEI)
Tarlatamab poate cauza SNCEI, inclusiv evenimente care pot pune viața în pericol sau pot fi letale.
În studiile clinice cu date privind siguranța cumulate de la 473 pacienți cu CPCM la care s-a administrat IMDYLLTRA în doză de 10 mg, SNCEI a fost raportat la 4,7% dintre pacienți. Timpul median de la prima doză de IMDYLLTRA până la primul debut al SNCEI a fost de 9,0 zile (interval intercuartilic: între 2 și 13 zile). Timpul median până la rezoluția SNCEI a fost de 4 zile (interval intercuartilic: între 2 și 8 zile).
Pentru abordarea terapeutică clinică a SNCEI, vezi pct. 4.4.
Neutropenia
În studiile clinice cu date privind siguranța cumulate de la 473 pacienți cu CPCM la care s-a administrat IMDYLLTRA în doză de 10 mg, neutropenia a apărut la 16,9% dintre pacienți, incluzând 8,2% dintre pacienții care au prezentat evenimente de gradul 3 sau gradul 4. Timpul median de la prima doză de IMDYLLTRA până la prima apariție a neutropeniei a fost de 43 zile (interval: între 29 și 109 zile). Neutropenia care a dus la întreruperea administrării dozei a apărut la 3,2% dintre pacienți, niciunul dintre aceste cazuri neducând la întreruperea tratamentului. Tratamentul cu FSCG a fost necesar la 6% dintre pacienți.
Pentru abordarea terapeutică clinică a neutropeniei, vezi pct. 4.4.
Raportarea reacțiilor adverse suspectate
Raportarea reacțiilor adverse suspectate după autorizarea medicamentului este importantă. Acest lucru permite monitorizarea continuă a raportului beneficiu/risc al medicamentului. Profesioniștii din domeniul sănătății sunt rugați să raporteze orice reacție adversă suspectată prin intermediul sistemului național de raportare, astfel cum este menționat în Anexa V.
4.9 Supradozaj
În studiile clinice au fost evaluate doze de până la 100 mg administrate la interval de 2 săptămâni și de 200 mg administrate la interval de 3 săptămâni. În caz de supradozaj, pacientul trebuie monitorizat atent pentru semne sau simptome de reacții adverse și trebuie tratat simptomatic și, după cum este necesar, trebuie instituite măsuri de susținere.
5. PROPRIETĂȚI FARMACOLOGICE
5.1 Proprietăți farmacodinamice
Grupa farmacoterapeutică: agenți antineoplazici, alți anticorpi monoclonali și conjugate anticorp-medicament, codul ATC: L01FX33
Mecanism de acțiune
Tarlatamab este un agent bispecific de angajare a celulelor T direcționat împotriva ligandului delta-like 3 (DLL3), care se leagă de DLL3 exprimat pe suprafața celulelor tumorale și de CD3 exprimat pe suprafața celulelor T. Tarlatamab se leagă bispecific de limfocitele T și de celulele tumorale care exprimă DLL3, determinând activarea limfocitelor T, producerea de citokine inflamatorii și eliberarea de proteine citotoxice, ceea ce conduce la liza direcționată a celulelor tumorale.
Efecte farmacodinamice
Răspunsul farmacodinamic după o singură perfuzie de tarlatamab a fost caracterizat prin redistribuirea și activarea celulelor T și creșterea tranzitorie a citokinelor. Redistribuirea periferică a celulelor T (adică aderența celulelor T la endoteliul vascular și/sau transmigrarea în țesut) a survenit în decurs de 24 ore de la administrarea dozei inițiale de tarlatamab de 1 mg din ziua 1. Numărul de celule T a scăzut în decurs de 6 ore de la perfuzie și a revenit la valorile inițiale la majoritatea pacienților, înainte de perfuzia următoare din ziua 8.
Valorile citokinelor serice IL-2, IL-6, IL-8, IL-10, IFN-γ și TNF-α au fost crescute tranzitoriu după administrarea dozei inițiale de tarlatamab de 1 mg din ziua 1. Citokinele au atins valori maxime în primele 2 zile de la începerea perfuziei cu tarlatamab și, în general, au revenit la valorile inițiale, înainte de perfuzia următoare în ziua 8. La tratamentele ulterioare, creșterea valorilor citokinelor a survenit la mai puțini pacienți, cu o intensitate mai redusă comparativ cu perfuzia inițială din ziua 1.
Imunogenitate
Anticorpii anti-medicament (ADA) au fost detectați frecvent. Nu s-au observat dovezi ale impactului ADA asupra farmacocineticii, eficacității sau siguranței; cu toate acestea, datele sunt încă limitate.
Eficacitate și siguranță clinică
Studiul DeLLphi-304
Eficacitatea IMDYLLTRA a fost studiată într-un studiu clinic de fază 3, multicentric randomizat deschis (Studiul DeLLphi-304). Pacienții eligibili trebuiau să fie diagnosticați cu CPCM cu progresie a bolii după 1 regim pe bază de platină. În regiunile în care tratamentul sistemic de primă linie, conform standardului de îngrijire (SOC) pentru pacienții diagnosticați cu boală în stadiu extins a inclus chimioterapie pe bază de platină în asociere cu un inhibitor al PD-(L)1, pacienții trebuiau fie să fi prezentat eșec la tratamentul cu inhibitor al PD-(L)1 ca parte a tratamentului sistemic de primă linie, fie să nu fie eligibili pentru terapia cu inhibitor al PD-(L)1. În plus, pacienții trebuiau să aibă statusul de performanță ECOG (Eastern Cooperative Oncology Group) 0-1 și cel puțin o leziune măsurabilă, conform criteriilor de evaluare a răspunsului în tumori solide (RECIST v1.1). Studiul a exclus pacienții cu metastaze cerebrale simptomatice sau cu imunodeficiență activă.
În total 509 pacienți au fost înrolați și randomizați 1:1 pentru a li se administra fie IMDYLLTRA, fie chimioterapie conform standardului de îngrijire (SOC). 254 pacienți au fost randomizați pentru a li se administra IMDYLLTRA în doză inițială de 1 mg în ziua 1 a Ciclului 1, urmată de o doză de 10 mg în zilele 8 și 15 și apoi la interval de 2 săptămâni, într-un ciclu de 28 zile, până la progresia bolii sau toxicitate inacceptabilă. Chimioterapiile SOC au inclus topotecan (n = 185), lurbinectedin (n = 47) sau amrubicină (n = 23). Randomizarea a fost stratificată în funcție de expunerea anterioară la anti-PD-(L)1 (da versus nu), statusul sensibilității la platină (interval fără chimioterapie ≥ 180 zile, < 180 până la ≥ 90 zile sau < 90 zile), prezența (anterioară sau curentă) a metastazelor cerebrale (da versus nu) și tratamentul administrat conform standardului de îngrijire (topotecan/amrubicină sau lurbinectedin). Tratamentul a continuat până la progresia bolii sau toxicitate inacceptabilă. Evaluările tumorale au fost efectuate la interval de 6 săptămâni în primele 48 săptămâni și apoi la interval de 12 săptămâni.
Caracteristicile demografice și ale bolii la momentul inițial ale populației din studiu au fost următoarele: vârstă mediană 65 ani (interval: 20-86 ani); 41,3% cu vârsta între 65 și 74 ani; 10,8% cu vârsta de 75 ani sau peste; 69% bărbați; 57,2% de rasă albă și 40,1% asiatici; 32% cu status de performanță ECOG 0 și 67,2% cu status de performanță ECOG 1; 91% dintre pacienți prezentau boală metastatică la momentul inițial; 44,8% prezentau metastaze cerebrale la momentul inițial; 35,2% prezentau metastaze hepatice la momentul inițial. 68,8% dintre pacienți erau foști fumători; 20,6% erau fumători, 10,6% nu fumaseră niciodată. Toți pacienții au fost tratați anterior cu cel puțin 1 linie de chimioterapie pe bază de platină (interval: 1 până la 3 linii); 97,6% dintre pacienți au fost tratați anterior cu 1 linie de tratament; 70,7% au fost tratați anterior cu terapie anti-PD-(L)1; 223 pacienți
(43,8%) avuseseră un interval fără chimioterapie < 90 zile ulterior încheierii terapiei de primă linie cu platină, în timp ce 286 pacienți (56,2%) avuseseră un interval fără chimioterapie ≥ 90 zile.
Principalul indicator al eficacității a fost supraviețuirea generală (SG). Criteriul cheie secundar de evaluare a eficacității a fost supraviețuirea fără progresia bolii (SFP) estimată pe baza evaluării investigatorului, conform criteriilor de evaluare a răspunsului în tumorile solide (RECIST v1.1) și pe baza anumitor rezultate raportate de pacienți. Criteriile finale suplimentare au inclus rata de răspuns generală (RRG) estimată pe baza evaluării investigatorului conform RECIST v1.1.
Pacienților li s-a administrat un număr median de 5 cicluri de tratament cu IMDYLLTRA (interval: 1 până la 19 cicluri) și un număr median de 4 cicluri de tratament SOC (interval: 1 până la 21 cicluri).
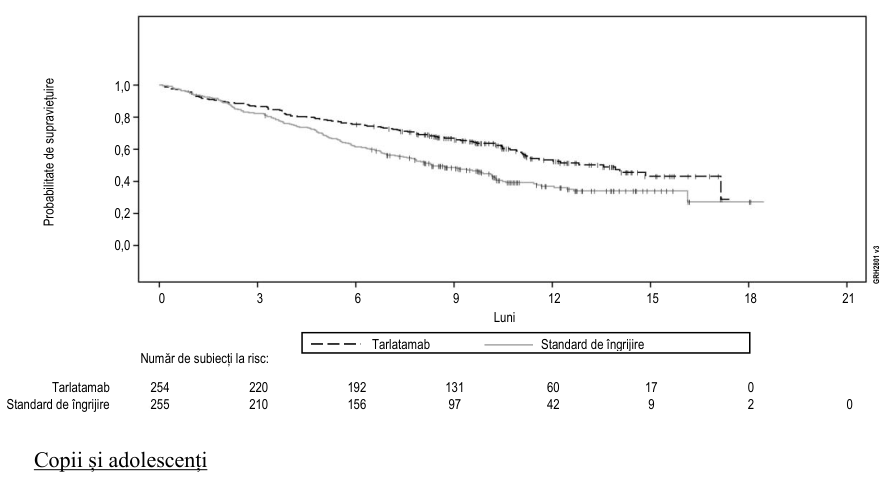
Rezultatele privind eficacitatea sunt rezumate în tabelul 9 și figura 1. Durata mediană de urmărire (IÎ 95%) pentru SG a fost de 11,2 luni (10,4, 12,1) în grupul tratat cu tarlatamab și de 11,7 luni (10,6, 12,3) în grupul tratat cu chimioterapie SOC. Durata mediană de urmărire (IÎ 95%) pentru SFP a fost de 11,0 luni (8,5, 11,2) pentru grupul tratat cu tarlatamab și de 9,7 luni (8,4, 11,1) pentru grupul tratat cu chimioterapie SOC.
Tabelul 9. Rezultatele privind eficacitatea pentru pacienții cu CPCM din Studiul DeLLphi-304
| Parametru de eficacitate | IMDYLLTRA (N = 254) | Standard de îngrijire (N = 255) |
|---|---|---|
| Supraviețuire generală (SG) | ||
| Decese (%) | 111 (43,7) | 152 (59,6) |
| Medianaa în luni (IÎ 95%) | 13,6 (11,1, NE) | 8,3 (7,0, 10,2) |
| Raport de riscb (IÎ 95%) | 0,60 (0,47, 0,77) | |
| Valoare p (test de rang logaritmic stratificat) | < 0,001 | |
| Supraviețuire fără progresia bolii (SFP)c | ||
| Evenimente (%) | 191 (75,2) | 205 (80,4) |
| Medianăa în luni (IÎ 95%) | 4,2 (3,0, 4,4) | 3,2 (2,9, 4,2) |
| Raport de riscb (IÎ 95%) | 0,72 (0,59, 0,88) | |
| Valoare p (test de rang logaritmic stratificat) | < 0,001 | |
| Rată de răspuns generală (RRG)c | ||
| RRG, % | 35,0 | 20,4 |
a Conform estimărilor Kaplan-Meier.
b Raport de risc bazat pe modelul Cox stratificat al riscurilor proporționale.
c SFP, RRG pe baza evaluării investigatorului conform RECIST v1.1.
IÎ = interval de încredere; N = număr; NE = nu poate fi estimat
Figura 1. Diagrama Kaplan-Meier a supraviețuirii generale (setul de analiză ITT)
Agenția Europeană pentru Medicamente a acordat o derogare de la obligația de depunere a rezultatelor studiilor efectuate cu tarlatamab la toate subgrupele de copii și adolescenți în tratamentul cancerului pulmonar cu celule mici (vezi pct. 4.2 pentru informații privind utilizarea la copii și adolescenți).
5.2 Proprietăți farmacocinetice
Analizele farmacocinetice (FC) populaționale ale tarlatamab la subiecți adulți (n = 702) cu CPCM avansat tratat anterior au fost efectuate pentru a caracteriza evoluția în timp a concentrațiilor serice de tarlatamab după administrarea intravenoasă, pentru a cuantifica variabilitatea interindividuală și pentru a evalua efectele covariabilelor specifice subiecților asupra parametrilor FC ai tarlatamab.
Concentrația serică maximă (Cmax), concentrația serică minimă (Cmin) și aria de sub curba concentrație serică-timp la starea de echilibru (ASCtau) ale tarlatamab au crescut proporțional cu doza în intervalul de doze evaluat, între 1 mg și 100 mg cu administrare la interval de 2 săptămâni (Q2S) (de 10 ori doza recomandată). Starea de echilibru aproximativă a expunerilor serice la tarlatamab a fost atinsă până în ziua 15 a ciclului 2.
Distribuție
Valoarea tipică (CV% între subiecți) pentru volumul central de distribuție este de 3,23 l (38%), iar volumul de distribuție la starea de echilibru este de 8,19 l, estimată prin analiza FC populațională.
Metabolizare
Calea metabolică a tarlatamabului nu a fost caracterizată. Similar altor terapii proteice, se așteaptă ca tarlatamab să fie degradat în peptide mici și aminoacizi prin căile catabolice.
Eliminare
Clearance-ul sistemic (CV% între subiecți) a fost de 0,728 l/zi (34%), iar timpul de înjumătățire plasmatică prin eliminare terminală a fost de aproximativ 10,6 zile la subiecții cu CPCM, conform estimării efectuate prin analiza FC populațională.
Grupe speciale de pacienți
Nu s-au observat diferențe semnificative clinic în clearance-ul tarlatamab în funcție de vârstă (interval: 20-86 ani), greutate corporală (interval: 35-149 kg), sex, rasă, insuficiență renală ușoară sau moderată (RFGe ≥ 30 ml/min) sau insuficiență hepatică ușoară (bilirubină totală ≤ limita superioară a valorilor normale (LSN) și AST > LSN). Sunt disponibile date limitate privind pacienții cu insuficiență hepatică moderată și nu sunt disponibile date privind pacienții cu insuficiență hepatică severă sau insuficiență renală severă.
5.3 Date preclinice de siguranță
Datele non-clinice nu au evidențiat niciun risc special pentru om pe baza studiilor convenționale de farmacologie de siguranță și toxicitate după doze repetate.
Genotoxicitate și carcinogenitate
Nu au fost efectuate studii de genotoxicitate sau carcinogenitate cu tarlatamab.
Afectarea fertilității
Nu au fost efectuate studii care să evalueze efectele tarlatamab asupra fertilității.
Toxicitate asupra aparatului reproducător și a dezvoltării
Un studiu cu privire la toxicitatea asupra aparatului reproducător efectuat la șoareci utilizând molecula surogat murină muS757 a arătat transportul transplacentar al muS757 și nu a indus toxicitate embrio-fetală sau teratogenitate.
6. PROPRIETĂȚI FARMACEUTICE
6.1 Lista excipienților
Pulbere
Acid glutamic
Sucroză
Polisorbat 80 (E433)
Hidroxid de sodiu (pentru ajustarea pH-ului)
Soluție (stabilizator)
Acid citric monohidrat (E330)
Clorhidrat de lizină
Polisorbat 80 (E433)
Hidroxid de sodiu (pentru ajustarea pH-ului)
Apă pentru preparate injectabile
6.2 Incompatibilități
Nu se cunosc incompatibilități.
6.3 Perioada de valabilitate
Flaconul sigilat
4 ani.
Soluția diluată pentru perfuzie intravenoasă (pungă de perfuzie)
Stabilitatea chimică și fizică în timpul utilizării a fost demonstrată pentru o durată de 28 zile de păstrare la temperaturi între 2 °C și 8 °C și de 8 ore la temperaturi între 20 °C și 25 °C.
Din punct de vedere microbiologic, medicamentul trebuie utilizat imediat. Dacă nu este utilizat imediat, timpii și condițiile de depozitare în vederea întrebuințării sunt responsabilitatea utilizatorului și în mod normal nu trebuie să depășească 24 ore la temperaturi între 2 °C și 8 °C, cu excepția cazului în care reconstituirea și diluarea au avut loc în condiții aseptice controlate și validate.
6.4 Precauții speciale pentru păstrare
A se păstra și transporta la frigider (între 2°C și 8°C).
A nu se congela.
A se păstra în ambalajul original pentru a fi protejat de lumină.
Pentru condițiile de păstrare ale medicamentului după reconstituire și diluare, vezi pct. 6.3.
6.5 Natura și conținutul ambalajului
IMDYLLTRA este disponibil în două configurații de ambalare. Fiecare ambalaj IMDYLLTRA conține 1 flacon de pulbere pentru concentrat pentru soluție perfuzabilă și 2 flacoane de soluție (stabilizator).
IMDYLLTRA 1 mg pulbere pentru concentrat și soluție pentru soluție perfuzabilă
- 1 mg de pulbere de tarlatamab într-un flacon de sticlă de tip 1, cu dop elastomeric, sigiliu din aluminiu și capac gri detașabil
- 7 ml de soluție într-un flacon de sticlă de tip 1, cu dop elastomeric, sigiliu din aluminiu și capac alb detașabil
IMDYLLTRA 10 mg pulbere pentru concentrat și soluție pentru soluție perfuzabilă
- 10 mg de pulbere de tarlatamab într-un flacon de sticlă de tip 1 cu dop elastomeric, sigiliu din aluminiu și capac portocaliu detașabil
- 7 ml de soluție într-un flacon de sticlă de tip 1 cu dop elastomeric, sigiliu din aluminiu și capac alb detașabil
6.6 Precauții speciale pentru eliminarea reziduurilor și alte instrucțiuni de manipulare
Preparare aseptică
Respectați cu strictețe tehnica aseptică la prepararea soluției pentru perfuzie, deoarece flacoanele de tarlatamab nu conțin conservanți antimicrobieni.
Alte instrucțiuni
- Reconstituirea IMDYLLTRA se face cu apă pentru preparate injectabile. Nu utilizați soluția (stabilizatorul) pentru a reconstitui IMDYLLTRA. Soluția (stabilizatorul) este utilizată pentru a amorsa punga de perfuzie înainte de a adăuga IMDYLLTRA reconstituit, pentru a preveni adsorbția IMDYLLTRA pe pereții pungii de perfuzie și ai liniei de perfuzare.
- Pungile de perfuzie fabricate din acetat de etil vinil (EVA), poliolefină și clorură de polivinil (PVC) s-au dovedit a fi compatibile cu tarlatamab în condițiile de administrare specificate.
- Materialele liniei de perfuzare și cateterului, fabricate din poliolefină, PVC și poliuretan, s-au dovedit a fi compatibile cu tarlatamabul în condițiile de administrare specificate.
- Utilizarea de dispozitive de transfer cu sistem închis (CSTD, Closed System Transfer Device) nu este recomandată, din cauza riscului potențial de eroare de medicație. Compatibilitatea dispozitivelor CSTD adaptoare de flacon cu IMDYLLTRA nu a fost studiată.
Prepararea soluției perfuzabile
Reconstituirea tarlatamab
Tabelul 10. Cantitatea de apă pentru preparate injectabile necesară pentru reconstituirea IMDYLLTRAa
Concentrația flaconului de IMDYLLTRA | Cantitatea de apă pentru preparate injectabile necesară pentru reconstituirea IMDYLLTRA | Concentrație finală |
|---|---|---|
| 1 mg | 1,3 ml | 0,9 mg/ml |
| 10 mg | 4,4 ml | 2,4 mg/ml |
a Fiecare flacon conține un volum suplimentar, pentru a permite extragerea a 1,1 ml (flacon de 1 mg) sau a 4,2 ml (flacon de 10 mg) după reconstituire, pentru a asigura administrarea în concentrația specificată pe eticheta flaconului.
- Transferați cantitatea necesară de apă pentru preparate injectabile (consultați tabelul 10) în flaconul de tarlatamab pentru a obține o concentrație finală de tarlatamab de 0,9 mg/ml (flacon de 1 mg) sau 2,4 mg/ml (flacon de 10 mg). Direcționați apa de-a lungul pereților flaconului de IMDYLLTRA, nu direct pe pulberea liofilizată.
- Nu utilizați soluția (stabilizatorul) pentru a reconstitui IMDYLLTRA.
- Rotiți ușor conținutul. Nu agitați.
- Inspectați vizual dacă soluția este limpede până la ușor opalescentă, incoloră până la ușor gălbuie. Nu utilizați soluția dacă este tulbure sau conține particule.
Pregătirea pungii de perfuzie IMDYLLTRA
Tabelul 11. Ghid de preparare pentru o perfuzie cu durata de 1 oră
| Concentrația flaconului de IMDYLLTRA | Doza de IMDYLLTRA | Volumul de soluție injectabilă de clorură de sodiu 9 mg/ml (0,9%) care trebuie extras din punga de perfuzie | Volumul de soluție (stabilizator) care trebuie adăugat în punga de perfuzie | Volumul de IMDYLLTRA reconstituit care trebuie adăugat în punga de perfuzie |
|---|---|---|---|---|
| 1 mg | 1 mg | 14 ml | 13 ml | 1,1 ml |
| 10 mg | 10 mg | 17 ml | 13 ml | 4,2 ml |
Notă: concentrațiile finale ale flacoanelor de concentrații diferite NU sunt aceleași după reconstituire.
- Utilizați o pungă de perfuzie preumplută cu 250 ml de soluție injectabilă de clorură de sodiu 9 mg/ml (0,9%).
- Aspirați volumul necesar de soluție injectabilă de clorură de sodiu 9 mg/ml (0,9%) din punga de perfuzie preumplută și aruncați-l (consultați tabelul 11). Ignorați orice volum suplimentar din punga de perfuzie.
- Adăugați soluție (stabilizator).
- Pentru a amorsa punga de perfuzie, transferați 13 ml de soluție (stabilizator) în punga de perfuzie care conține soluție injectabilă de clorură de sodiu 9 mg/ml (0,9%).
- Omogenizați conținutul pungii cu mișcări ușoare, pentru a evita formarea de spumă. Nu agitați.
- Adăugați IMDYLLTRA reconstituit.
- Transferați volumul necesar de IMDYLLTRA reconstituit în punga de perfuzie stabilizată care conține soluție injectabilă de clorură de sodiu 9 mg/ml (0,9%) și soluția (stabilizatorul). Consultați tabelul 11.
- Omogenizați conținutul pungii cu mișcări ușoare, pentru a evita formarea de spumă. Nu agitați.
- Evacuați aerul din punga de perfuzie folosind o seringă goală, pentru a evita formarea de spumă.
- Amorsați linia de perfuzie cu soluție injectabilă de clorură de sodiu 9 mg/ml (0,9%) sau cu medicamentul final preparat din punga de perfuzie.
Timpul de depozitare conform pct. 6.3 include timpul total permis de la momentul reconstituirii primului flacon până la sfârșitul administrării. După scoaterea din frigider, lăsați punga de perfuzie să ajungă la temperatura camerei și finalizați administrarea soluției perfuzabile diluate de IMDYLLTRA în timpul permis în condițiile de păstrare la temperatura camerei (luând în calcul inclusiv timpul de perfuzare). Dacă punga de perfuzie cu tarlatamab pregătită nu este administrată în intervalele de timp și la temperaturile indicate, aceasta trebuie aruncată; nu trebuie introdusă din nou la frigider.
Orice medicament neutilizat sau material rezidual trebuie eliminat în conformitate cu reglementările locale.
7. DEȚINĂTORUL AUTORIZAȚIEI DE PUNERE PE PIAȚĂ
Amgen Europe B.V.
Minervum 7061
4817 ZK Breda
Olanda
8. NUMĂRUL(ELE) AUTORIZAȚIEI DE PUNERE PE PIAȚĂ
EU/1/26/2033/001
EU/1/26/2033/002
9. DATA PRIMEI AUTORIZĂRI SAU A REÎNNOIRII AUTORIZAȚIEI
Data primei autorizări:
10. DATA REVIZUIRII TEXTULUI
Informații detaliate privind acest medicament sunt disponibile pe site-ul Agenției Europene pentru Medicamente https://www.ema.europa.eu.