RYBREVANT 1600 mg
Rezumatul caracteristicilor produsului (RCP)
- 1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI
- 2. COMPOZIȚIA CALITATIVǍ ȘI CANTITATIVǍ
- 3. FORMA FARMACEUTICĂ
-
4. DATE CLINICE
- 4.1 Indicații terapeutice
- 4.2 Doze și mod de administrare
- 4.3 Contraindicații
- 4.4 Atenționări și precauții speciale pentru utilizare
- 4.5 Interacțiuni cu alte medicamente și alte forme de interacțiune
- 4.6 Fertilitatea, sarcina și alăptarea
- 4.7 Efecte asupra capacității de a conduce vehicule și de a folosi utilaje
- 4.8 Reacții adverse
- 4.9 Supradozaj
- 5. PROPRIETĂȚI FARMACOLOGICE
- 6. PROPRIETĂȚI FARMACEUTICE
- 7. DEȚINĂTORUL AUTORIZAȚIEI DE PUNERE PE PIAȚĂ
- 8. NUMĂRUL(ELE) AUTORIZAȚIEI DE PUNERE PE PIAȚĂ
- 9. DATA PRIMEI AUTORIZĂRI SAU A REÎNNOIRII AUTORIZAȚIEI
- 10. DATA REVIZUIRII TEXTULUI
- 1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI
- 2. COMPOZIȚIA CALITATIVǍ ȘI CANTITATIVǍ
- 3. FORMA FARMACEUTICĂ
-
4. DATE CLINICE
- 4.1 Indicații terapeutice
- 4.2 Doze și mod de administrare
- 4.3 Contraindicații
- 4.4 Atenționări și precauții speciale pentru utilizare
- 4.5 Interacțiuni cu alte medicamente și alte forme de interacțiune
- 4.6 Fertilitatea, sarcina și alăptarea
- 4.7 Efecte asupra capacității de a conduce vehicule și de a folosi utilaje
- 4.8 Reacții adverse
- 4.9 Supradozaj
- 5. PROPRIETĂȚI FARMACOLOGICE
- 6. PROPRIETĂȚI FARMACEUTICE
- 7. DEȚINĂTORUL AUTORIZAȚIEI DE PUNERE PE PIAȚĂ
- 8. NUMĂRUL(ELE) AUTORIZAȚIEI DE PUNERE PE PIAȚĂ
- 9. DATA PRIMEI AUTORIZĂRI SAU A REÎNNOIRII AUTORIZAȚIEI
- 10. DATA REVIZUIRII TEXTULUI
1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI
Rybrevant 350 mg concentrat pentru soluție perfuzabilă
2. COMPOZIȚIA CALITATIVǍ ȘI CANTITATIVǍ
Un ml de concentrat pentru soluție perfuzabilă conține amivantamab 50 mg.
Un flacon de 7 ml conține 350 mg de amivantamab.
Amivantamab este un anticorp bispecific complet uman, pe bază de imunoglobulină G1 (IgG1), care vizează factorul de creștere epidermică (EGF) și receptorii de tranziție mezenchimal-epidermică (TME), produși de o linie de celule ale mamiferelor (ovar de hamster chinezesc [CHO]) utilizând tehnologia ADN-ului recombinant.
Excipienți cu efect cunoscut:
Un ml de soluție conține 0,6 mg de polisorbat 80.
Pentru lista tuturor excipienților, vezi pct. 6.1.
3. FORMA FARMACEUTICĂ
Concentrat pentru soluție perfuzabilă.
Soluția este de la incoloră până la galben deschis, are un pH de 5,7 și osmolalitate de aproximativ 310 mOsm/Kg.
4. DATE CLINICE
4.1 Indicații terapeutice
Rybrevant este indicat:
- în asociere cu lazertinib pentru tratamentul de primă linie la pacienții adulți cu cancer pulmonar fără celule mici (NSCLC) în stadiu avansat, cu deleții în Exonul 19 al EGFR sau cu mutații de substituție în Exonul 21 L858R.
- în asociere cu carboplatină și pemetrexed pentru tratamentul pacienților adulți cu NSCLC în stadiu avansat, cu deleții în Exonul 19 al EGFR sau cu mutații de substituție în Exonul 21 L858R, după eșecul tratamentului anterior care a inclus un inhibitor de tirozin kinază (TKI) al EGFR.
- în asociere cu carboplatină și pemetrexed pentru tratamentul de primă linie la pacienții adulți cu NSCLC în stadiu avansat, cu mutații activatoare ale inserției Exon 20 a EGFR.
- în monoterapie, pentru tratamentul pacienților adulți cu cancer pulmonar fără celule mici (NSCLC) în stadiu avansat, cu mutații activatoare ale inserției Exon 20 a EGFR, după eșecul terapiei pe bază de platină.
4.2 Doze și mod de administrare
Tratamentul cu Rybrevant trebuie inițiat și supervizat de un medic cu experiență în utilizarea medicamentelor antineoplazice.
Rybrevant trebuie administrat de către un profesionist din domeniul sănătății, care să aibă acces la asistență medicală adecvată pentru abordarea terapeutică a reacțiilor adverse legate de perfuzie (RALP-uri), dacă apar.
Înaintea inițierii tratamentului cu Rybrevant, trebuie determinată existența mutației EGFR în eșantioanele de țesut tumoral sau plasmatic utilizând o metodă de testare validată. Dacă nu se detectează nicio mutație în eșantionul plasmatic, trebuie testat dacă țesutul tumoral este disponibil întro cantitate suficientă și are o calitate adecvată, din cauza potențialului de rezultate fals-negative ale unui test bazat pe plasmă. Testarea poate fi efectuată în orice moment de la diagnosticul inițial până la inițierea tratamentului; nu este necesară repetarea testării odată ce statusul mutației EGFR a fost stabilit (vezi pct. 5.1).
Doze
Trebuie administrate medicații prealabile pentru a reduce riscul de RALP-uri asociate cu Rybrevant (vezi mai jos „Ajustările dozei” și „Medicația concomitentă recomandată”).
La fiecare 3 săptămâni
Dozele recomandate de Rybrevant, atunci când este utilizat în asociere cu carboplatină și pemetrexed, sunt prezentate în Tabelul 1 (vezi mai jos „Vitezele de perfuzare” și Tabelul 5).
Tabelul 1: Doza recomandată de Rybrevant la fiecare 3 săptămâni
| Greutatea corporală a pacientului la momentul inițiala | Doza de Rybrevant | Schemă | Număr de flacoane |
|---|---|---|---|
| Mai mică de 80 kg | 1400 mg | Săptămânal (în total 4 doze) din săptămâna 1 până în săptămâna 4
| 4 |
| 1750 mg | La fiecare 3 săptămâni, începând cu săptămâna 7 | 5 | |
| Mai mare sau egală cu 80 kg | 1750 mg | Săptămânal (în total 4 doze) – săptămânile 1 până la 4
| 5 |
| 2100 mg | La fiecare 3 săptămâni, începând cu săptămâna 7 | 6 |
a
Ajustările dozei nu sunt necesare pentru modificările ulterioare ale greutății corporale.
Atunci când se utilizează în asociere cu carboplatină și pemetrexed, Rybrevant trebuie administrat după carboplatină și pemetrexed în următoarea ordine: pemetrexed, carboplatină și apoi Rybrevant. Pentru instrucțiunile de dozare pentru carboplatină și pemetrexed, vezi pct. 5.1 și informațiile de prescriere ale fabricantului.
La fiecare 2 săptămâni
Dozele recomandate de Rybrevant în monoterapie sau în asociere cu lazertinib sunt prezentate în Tabelul 2 (vezi mai jos „Vitezele de perfuzare” și Tabelul 6).
Tabelul 2: Doză recomandată de Rybrevant la fiecare 2 săptămâni
| Greutatea corporală a pacientului la momentul inițiala | Doza de Rybrevant | Schemă | Număr de flacoane de Rybrevant 350 mg/7 ml |
|---|---|---|---|
| Mai mică de 80 kg | 1050 mg | Săptămânal (în total 4 doze) din săptămâna 1 până în săptămâna 4
| 3 |
| La fiecare 2 săptămâni, începând cu săptămâna 5 | |||
| Mai mare sau egală cu 80 kg | 1400 mg | Săptămânal (în total 4 doze) – săptămânile 1 până la 4
| 4 |
| La fiecare 2 săptămâni, începând cu săptămâna 5 |
a
Ajustările dozei nu sunt necesare pentru modificările ulterioare ale greutății corporale.
Dacă se administrează în asociere cu lazertinib, se recomandă administrarea Rybrevant în orice moment după administrarea lazertinib, dacă sunt adminstrate în aceeași zi. Consultați pct. 4.2 din Rezumatul caracteristicilor produsului pentru lazertinib pentru informațiile privind dozele recomandate de lazertinib.
Durata tratamentului
Se recomandă ca pacienților să li de administreze Rybrevant până la progresia bolii sau până la apariția toxicității inacceptabile.
Doza omisă
Dacă se omite o doză planificată, doza trebuie administrată cât mai curând posibil, iar schema de administrare trebuie ajustată în consecință, menținând intervalul de tratament.
Ajustările dozei
În cazul reacțiilor adverse de grad 3 sau 4, administrarea trebuie întreruptă până la momentul ameliorării reacțiilor adverse până la reacții adverse de grad ≤ 1 sau revenirea la starea inițială. Dacă o întrerupere durează 7 zile sau mai puțin, reîncepeți cu doza curentă. Dacă o întrerupere durează mai mult de 7 zile, se recomandă reînceperea tratamentului cu o doză redusă, așa cum este prezentat în Tabelul 3. După Tabelul 3 sunt prezentate, de asemenea, și modificările specifice ale dozei în funcție de reacțiile adverse specifice.
Dacă se administrează în asociere cu lazertinib, consultați pct. 4.2 din Rezumatul caracteristicilor produsului pentru lazertinib pentru informații privind ajustările dozei.
Tabelul 3: Recomandări privind modificarea dozei în cazul apariției reacțiilor adverse
| Doza la care au apărut reacțiile adverse | Doza după prima întrerupere determinată de apariția reacțiilor adverse | Doza după a doua întrerupere determinată de apariția reacțiilor adverse | Doză după a treia întrerupere determinată de apariția reacțiilor adverse |
|---|---|---|---|
| 1050 mg | 700 mg | 350 mg | Se oprește tratamentul cu Rybrevant |
| 1400 mg | 1050 mg | 700 mg | |
| 1750 mg | 1400 mg | 1050 mg | |
| 2100 mg | 1750 mg | 1400 mg |
Abordarea terapeutică a reacțiilor adverse legate de perfuzie
Perfuzia trebuie întreruptă la primul semn de RALP. Tratamente suplimentare de susținere a funcțiilor vitale (de exemplu, glucocorticoizi i, antihistaminice, antipiretice și antiemetice suplimentare) trebuie administrate conform indicațiilor clinice (vezi pct. 4.4).
- Gradul 1-3 (ușor-sever): Odată cu recuperarea în urma simptomelor, se reia perfuzia cu 50% din viteza anterioară. Dacă nu există alte simptome, viteza de perfuzare poate fi crescută în funcție de viteza de perfuzare recomandată (vezi Tabelul 5 și Tabelul 6). Medicamentele administrate concomitent trebuie administrate cu următoarea doză [inclusiv dexametazonă (20 mg) sau echivalent] (vezi Tabelul 4).
- Recidivă de grad 3 sau grad 4 (cu potențial letal): Întrerupeți definitiv tratamentul cu Rybrevant.
Evenimente tromboembolice venoase (TEV) asociate cu administrarea în asociere cu lazertinib La inițierea tratamentului, trebuie administrate profilactic anticoagulante, pentru a preveni evenimentele TEV la pacienții cărora li se administrează Rybrevant în asociere cu lazertinib. Conform ghidurilor clinice, pacienților trebuie să li se administreze tratament profilactic fie cu un anticoagulant oral cu acțiune directă (AOAD), fie cu o heparină cu masă moleculară mică (HMMM). Utilizarea antagoniștilor vitaminei K nu este recomandată.
Pentru evenimentele TEV asociate cu instabilitate clinică (de ex. insuficiență respiratorie sau disfuncție cardiacă), administrarea ambelor medicamente trebuie oprită până când pacientul este stabil clinic. După aceea, administrarea ambelor medicamente poate fi reluată în aceleași doze. În cazul recurenței, în ciuda tratamentului adecvat cu anticoagulant, se întrerupe administrarea Rybrevant. Tratamentul poate continua cu lazertinib în aceeași doză.
Reacții cutanate și unghiale
Se recomandă terapia profilactică cu antibiotice cu administrare orală și topică pentru a reduce riscul de apariție și severitatea reacțiilor la nivelul pielii și unghiilor la pacienții cărora li se administrează Rybrevant. Se recomandă, de asemenea, utilizarea unei creme hidratante necomedogene (de preferință pe bază de ceramide sau alte formule care asigură hidratarea îndelungată a pielii și nu conțin agenți de uscare) pe față și pe întreg corpul (cu excepția scalpului) și a unei soluții de clorhexidină pentru spălarea mâinilor și a picioarelor. Pacienții trebuie sfătuiți să limiteze expunerea la soare pe durata tratamentului cu Rybrevant și timp de 2 luni după acesta. Pentru informații suplimentare despre profilaxia reacțiilor cutanate și unghiale, vezi pct. 4.4.
Dacă pacientul dezvoltă o reacție cutanată sau unghială de grad 1-2, trebuie inițiată terapia de susținere, conform indicațiilor clinice; dacă nu există nicio ameliorare după 2 săptămâni, pentru erupția cutanată persistentă de gradul 2, trebuie luată în considerare reducerea dozei (vezi Tabelul 3). Dacă pacientul dezvoltă o reacție cutanată sau unghială de grad 3, trebuie inițiat tratamentul de susținere, conform indicațiilor clinice și trebuie luată în considerare întreruperea tratamentului cu Rybrevant până la ameliorarea reacției adverse. După dispariția reacției cutanate sau unghiale ≤grad 2, tratamentul cu Rybrevant trebuie reluat cu o doză redusă. Dacă pacientul dezvoltă reacții cutanate de gradul 4, tratamentul cu Rybrevant trebuie întrerupt permanent (vezi pct. 4.4).
Boala pulmonară interstițială
Tratamentul cu Rybrevant trebuie oprit dacă se suspectează boală pulmonară interstițială (BPI) sau reacții adverse asemănătoare BPI (pneumonită). Dacă se confirmă că pacientul a dezvoltat BPI sau reacții adverse similare BPI (de exemplu, pneumonită), tratamentul cu Rybrevant trebuie oprit permanent (vezi pct. 4.4).
Medicația concomitentă recomandată
Cu două zile înaintea primei perfuzii:
În decursul celor două zile înainte de perfuzia inițială cu Rybrevant, pacienților trebuie să li se administreze 8 mg dexametazonă pe cale orală, de două ori pe zi.
În ziua perfuziei:
În ziua perfuziei inițiale (săptămâna 1, ziua 1), pacienților trebuie să li se administreze 8 mg dexametazonă pe cale orală, cu o oră înainte de perfuzie, suplimentar la dexametazona administrată intravenos, pentru a reduce și mai mult riscul de RALP-uri.
Înainte de perfuzie (săptămâna 1, zilele 1 și 2), trebuie administrate antihistaminice, antipiretice și glucocorticoizi pentru a reduce riscul de RALP-uri (vezi Tabelul 4). Pentru dozele ulterioare, este necesară administrarea de antihistaminice și antipiretice. După o perioadă prelungită de întrerupere a administrării dozei, trebuie reluată de asemenea administrarea de glucocorticoizi. Trebuie administrate antiemetice, dacă este necesar.
Tabelul 4: Schema de administrare a premedicației
| Medicație prealabilă | Doză | Cale de administrare | Interval recomandat de administrare înainte de administrarea Rybrevant |
|---|---|---|---|
| Antihistaminic* | Difenhidramină (25 până la 50 mg) sau echivalent | Intravenoasă | 15 până la 30 de minute |
| Orală | 30 până la 60 de minute | ||
| Antipiretic* | Paracetamol/Acetaminofen (650 până la 1000 mg) | Intravenoasă | 15 până la 30 de minute |
| Orală | 30 până la 60 de minute | ||
| Glucocorticoid ‡ | Dexametazonă (8 mg) | Orală | 60 de minute |
| Glucocorticoid‡ | Dexametazonă (20 mg) sau echivalent | Intravenoasă | 60 până la 120 de minute |
| Glucocorticoid+ | Dexametazonă (10 mg) sau echivalent | Intravenoasă | 45 până la 60 de minute |
* Necesar la toate dozele.
‡ Necesar la doza inițială (săptămâna 1, ziua 1) sau la următoarea doză ulterioară în cazul unei RALP
+ Necesar la a doua doză (săptămâna 1, ziua 2); opțional pentru dozele ulterioare.
Categorii speciale de populație
Copii și adolescenți
Utilizarea amivantamab nu se justifică la copii și adolescenți în tratamentul cancerului pulmonar fără celule mici.
Vârstnici
Nu sunt necesare ajustări ale dozei (vezi pct. 4.8, pct. 5.1 și pct. 5.2).
Insuficiență renală
Nu s-au efectuat studii specifice cu amivantamab la pacienți cu insuficiență renală. Pe baza analizelor de farmacocinetică (FC) populațională, nu este necesară ajustarea dozei la pacienții cu insuficiență renală ușoară sau moderată. Este necesară prudență la pacienții cu insuficiență renală severă, deoarece amivantamabul nu a fost studiat la această grupă de pacienți (vezi pct. 5.2). În cazul inițierii tratamentului, pacienții trebuie monitorizați pentru depistarea reacțiilor adverse, cu modificări ale dozei, conform recomandărilor de mai sus.
Insuficiență hepatică
Nu s-au efectuat studii specifice cu amivantamab la pacienți cu insuficiență hepatică. Pe baza analizelor de farmacocinetică populațională, nu este necesară ajustarea dozei la pacienții cu insuficiență hepatică ușoară. Este necesară prudență la pacienții cu insuficiență hepatică moderată sau severă, deoarece amivantamab nu a fost studiat la această grupă de pacienți (vezi pct. 5.2). În cazul inițierii tratamentului, pacienții trebuie monitorizați pentru apariția reacțiilor adverse, cu ajustări ale dozei, conform recomandărilor de mai sus.
Mod de administrare
Rybrevant este destinat administrării intravenoase. Se administrează sub formă de perfuzie intravenoasă după diluarea cu soluție injectabilă sterilă de glucoză 5% sau soluție injectabilă de clorură de sodiu 9 mg/ml (0,9%). Rybrevant trebuie administrat cu un filtru inclus în linia de perfuzie.
Pentru instrucțiuni privind diluarea medicamentului înainte de administrare, vezi pct. 6.6.
Vitezele de perfuzare
După diluare, perfuzia trebuie administrată intravenos la vitezele de perfuzare prezentate în Tabelul 5 sau în Tabelul 6 de mai jos. Din cauza frecvenței RALP-urilor la prima doză, amivantamab trebuie perfuzat în vena periferică în săptămâna 1 și în săptămâna 2; perfuzia cu o linie centrală poate fi administrată în săptămânile următoare, când riscul de RALP este mai mic (vezi pct. 6.6). Se recomandă ca prima doză să fie preparată cât mai aproape posibil de administrare, pentru a maximiza probabilitatea terminării perfuziei în cazul unei RALP.
Tabelul 5: Vitezele de perfuzare pentru administrarea Rybrevant la fiecare 3 săptămâni
| Greutate corporală mai mică de 80 kg | |||
|---|---|---|---|
| Săptămână | Doză (per pungă de 250 ml) | Viteza de perfuzare inițială | Viteza de perfuzare ulterioarㆠ|
| Săptămâna 1 (perfuzare cu doză împărțită) | |||
| Săptămâna 1 Ziua 1 | 350 mg | 50 ml/h | 75 ml/h |
| Săptămâna 1 Ziua 2 | 1050 mg | 33 ml/h | 50 ml/h |
| Săptămâna 2 | 1400 mg | 65 ml/h | |
| Săptămâna 3 | 1400 mg | 85 ml/h | |
| Săptămâna 4 | 1400 mg | 125 ml/h | |
| Săptămânile ulterioare* | 1750 mg | 125 ml/h | |
| Greutate corporală mai mare sau egală cu 80 kg | |||
| Săptămână | Doză (per pungă de 250 ml) | Viteza de perfuzare inițială | Viteza de perfuzare ulterioarㆠ|
| Săptămâna 1 (perfuzare cu doză împărțită) | |||
| Săptămâna 1 Ziua 1 | 350 mg | 50 ml/h | 75 ml/h |
| Săptămâna 1 Ziua 2 | 1400 mg | 25 ml/h | 50 ml/h |
| Săptămâna 2 | 1750 mg | 65 ml/h | |
| Săptămâna 3 | 1750 mg | 85 ml/h | |
| Săptămâna 4 | 1750 mg | 125 ml/h | |
| Săptămânile ulterioare* | 2100 mg | 125 ml/h | |
* Începând cu săptămâna 7, se administrează pacienților la fiecare 3 săptămâni.
† Creșteți viteza de perfuzare inițială la viteza de perfuzare ulterioară după 2 ore, în absența RALP-urilor.
Tabelul 6: Vitezele de perfuzare pentru administrarea Rybrevant la fiecare 2 săptămâni
| Greutate corporală mai mică de 80 kg | |||
|---|---|---|---|
| Săptămână | Doză (per pungă de 250 ml) | Viteza de perfuzare inițială | Viteza de perfuzare ulterioar㇠|
| Săptămâna 1 (perfuzare cu doză împărțită) | |||
| Săptămâna 1 Ziua 1 | 350 mg | 50 ml/h | 75 ml/h |
| Săptămâna 1 Ziua 2 | 700 mg | 50 ml/h | 75 ml/h |
| Săptămâna 2 | 1050 mg | 85 ml/h | |
| Săptămânile ulterioare* | 1050 mg | 125 ml/h | |
| Greutate corporală mai mare sau egală cu 80 kg | |||
| Săptămână | Doză (per pungă de 250 ml) | Viteza de perfuzare inițială | Viteza de perfuzare ulterioar㇠|
| Săptămâna 1 (perfuzare cu doză împărțită) | |||
| Săptămâna 1 Ziua 1 | 350 mg | 50 ml/h | 75 ml/h |
| Săptămâna 1 Ziua 2 | 1050 mg | 35 ml/h | 50 ml/h |
| Săptămâna 2 | 1400 mg | 65 ml/h | |
| Săptămâna 3 | 1400 mg | 85 ml/h | |
| Săptămânile ulterioare* | 1400 mg | 125 ml/h | |
* După săptămâna 5, se administrează pacienților la fiecare 2 săptămâni.
‡ Creșteți viteza de perfuzare inițială la viteza de perfuzare ulterioară după 2 ore, în absența RALP-urilor.
4.3 Contraindicații
Hipersensibilitate la substanța(ele) activă(e) sau la oricare dintre excipienții enumerați la pct. 6.1.
4.4 Atenționări și precauții speciale pentru utilizare
Trasabilitate
Pentru a avea sub control trasabilitatea medicamentelor biologice, numele și numărul lotului medicamentului administrat trebuie înregistrate cu atenție.
Reacții adverse legate de perfuzie
Reacțiile adverse legate de perfuzie au apărut frecvent la pacienții tratați cu amivantamab (vezi pct. 4.8).
Înainte de perfuzia inițială (Săptămâna 1), trebuie administrate antihistaminice, antipiretice și glucocorticoizi pentru a reduce riscul de RALP-uri. Pentru dozele ulterioare, trebuie administrate antihistaminice și antipiretice. Perfuzia inițială trebuie administrată în doze divizate în Săptămâna 1, Ziua 1 și 2.
Pacienții trebuie tratați în unități medicale adecvate abordului terapeutic al RALP. Perfuziile trebuie întrerupte la primul semn de RALP-uri de orice severitate, iar medicamentele administrate după perfuzie trebuie administrate conform indicațiilor clinice. După remisiunea simptomelor, perfuzia trebuie reluată la 50% din viteza anterioară. În cazul RALP-urilor recurente de grad 3 sau grad 4, tratamentul cu Rybrevant trebuie întrerupt permanent (vezi pct. 4.2).
Boala pulmonară interstițială
La pacienții cărora li se administrează amivantamab au fost raportate boli pulmonare interstițiale (BPI) sau reacții adverse similare BPI (de exemplu, pneumonită), inclusiv evenimente cu evoluție letală (vezi pct. 4.8). Pacienții trebuie monitorizați pentru simptome care indică BPI/pneumonită (de exemplu, dispnee, tuse, febră). Dacă apar simptome, tratamentul cu Rybrevant trebuie întrerupt până la evaluarea acestor simptome. Trebuie evaluată BPI suspectată sau reacțiile adverse asemănătoare BPI și, dacă este cazul, trebuie inițiat tratamentul adecvat. Rybrevant trebuie întrerupt definitiv la pacienții cu BPI confirmată sau reacții adverse asemănătoare BPI (vezi pct. 4.2).
Evenimente tromboembolice venoase (TEV) asociate cu administrarea în asociere cu lazertinib La pacienții cărora li s-a administrat Rybrevant în asociere cu lazertinib au fost raportate evenimente tromboembolice venoase (TEV), inclusiv tromboză venoasă profundă (TVP) și embolie pulmonară (EP) și evenimente cu evoluție letală (vezi pct. 4.8). Evenimentele TEV au apărut predominant în primele patru luni de tratament. Ar trebui administrate profilactic anticoagulante în primele patru luni de tratament, pentru a preveni TEV. Conform ghidurilor clinice, pacienților trebuie să li se administreze preventiv fie un anticoagulant oral cu acțiune directă (AOAD), fie o heparină cu masă moleculară mică (HMMM). Utilizarea antagoniștilor vitaminei K nu este recomandată.
Semnele și simptomele de evenimente TEV trebuie monitorizate. Pacienții cu evenimente TEV trebuie tratați cu anticoagulante după cum este indicat clinic. Pentru evenimentele TEV asociate cu instabilitate clinică, tratamentul trebuie oprit până când pacientul este stabil clinic. După aceea, ambele medicamente pot fi reluate în aceeași doză.
În cazul recurenței apărute în ciuda tratamentului adecvat cu anticoagulant, tratamentul cu Rybrevant trebuie oprit definitiv. Tratamentul poate continua cu lazertinib în aceeași doză (vezi pct. 4.2).
Reacții cutanate și unghiale
La pacienții cărora li se administrează amivantamab au apărut erupții cutanate (inclusiv dermatită acneiformă), prurit, xerodermie și ulcerații cutanate (vezi pct. 4.8). Pacienții trebuie instruiți să limiteze expunerea la soare în timpul și timp de 2 luni după tratamentul cu Rybrevant. Se recomandă echipament de protecție și utilizarea de creme cu factor protecție solară cu spectru larg UVA/UVB. Este recomandată abordare profilactică pentru prevenirea erupțiilor cutanate. Aceasta include terapie profilactică, la inițierea tratamentului, cu un antibiotic cu utilizare orală (de ex. doxiciclină sau minociclină, 100 mg, de două ori pe zi) începând cu Ziua 1, în primele 12 săptămâni de tratament, iar după finalizarea terapiei cu antibiotic cu utilizare orală, aplicarea topică a unei loțiuni cu antibiotic pe scalp (de ex. clindamicină 1%) pentru următoarele 9 luni de tratament. Este recomandată utilizarea unei creme hidratante necomedogene (sunt preferate formulele pe bază de ceramide sau alte formule care asigură hidratarea îndelungată a pielii și nu conțin agenți de uscare) pe față și pe întregul corp (cu excepția scalpului) și a unei soluții de clorhexidină pentru spălarea mâinilor și a picioarelor începând în Ziua 1 și continuând pe toată durata tratamentului.
Se recomandă ca, la momentul administrării dozei inițiale, să fie disponibile prescripții pentru antibiotice de uz topic și/sau cu administrare orală și corticosteroizi de uz topic pentru a reduce la minimum orice întârziere în abordarea terapeutică a erupției cutanate tranzitorii, în cazul în care aceasta se manifestă în ciuda tratamentului profilactic. Dacă apar reacții cutanate, trebuie administrată terapie de susținere, precum și corticosteroizi topici și antibiotice cu utilizare topică și/sau orală. În cazul evenimentelor de grad 3 sau al celor slab tolerate de grad 2, trebuie administrate, de asemenea, antibiotice sistemice și corticosteroizi cu administrare orală. Pacienții care prezintă erupții cutanate severe care au un aspect sau o distribuție atipică sau care nu prezintă o ameliorare în decurs de 2 săptămâni trebuie să se adreseze imediat unui dermatolog. Rybrevant trebuie redus, întrerupt sau întrerupt permanent în funcție de severitate (vezi pct. 4.2).
S-a raportat apariția necrolizei epidermice toxice (NET). Tratamentul cu acest medicament trebuie întrerupt dacă se confirmă NET.
Tulburări oculare
La pacienții cărora li se administrează amivantamab au apărut tulburări oculare, inclusiv cheratită (vezi pct. 4.8). Pacienții care prezintă agravarea simptomelor oculare trebuie îndrumați imediat către un oftalmolog și trebuie să întrerupă utilizarea lentilelor de contact până la evaluarea simptomelor. Vezi pct. 4.2 pentru modificările de doză în cazul reacțiilor adverse oculare de grad 3 sau 4.
Conținutul de sodiu
Acest medicament conține mai puțin de 1 mmol (23 mg) de sodiu per doză, adică practic „nu conține sodiu“. Pentru a prepara soluția perfuzabilă, acest medicament trebuie diluat în soluție de clorură de sodiu 9 mg/ml (0,9%). Acest aspect trebuie avut în vedere în cazul pacienților care urmează o dietă cu restricție sodată (vezi pct. 6.6).
Conținutul de polisorbat
Acest medicament conține 0,6 mg de polisorbat 80 în fiecare ml, echivalent cu 4,2 mg per flacon de 7 ml. Polisorbații pot cauza reacții de hipersensibilitate.
4.5 Interacțiuni cu alte medicamente și alte forme de interacțiune
Nu s-au efectuat studii privind interacțiunile cu alte medicamente. Ca anticorp monoclonal IgG1, este puțin probabil ca excreția renală și metabolizarea mediată prin intermediul enzimelor hepatice a amivantamabului nemodificat să fie căi majore de eliminare. Ca atare, nu se așteaptă ca variațiile enzimelor de metabolizare a medicamentului să afecteze eliminarea amivantamab. Datorită afinității mari față de un epitop unic al EGFR și TME, nu se anticipează ca amivantamab să modifice enzimele care metabolizează medicamentul.
Vaccinare
Nu există informații clinice disponibile legate de eficacitatea și profilul de siguranță ale administrării vaccinurilor la pacienții cărora li se administrează amivantamab. Evitați administrarea de vaccinuri cu virusuri vii sau cu virusuri vii-atenuate la pacienții cărora li se administrează amivantamab.
4.6 Fertilitatea, sarcina și alăptarea
Femeile aflate la vârsta fertilă/Contracepția
Femeile aflate la vârsta fertilă trebuie să utilizeze măsuri contraceptive eficace în timpul și până la 3 luni după întreruperea tratamentului cu amivantamab.
Sarcina
Nu există date la om pentru a evalua riscul utilizării amivantamab în timpul sarcinii. Nu au fost efectuate studii de reproducere pe animale pentru a identifica un risc asociat medicamentului. Administrarea moleculelor inhibitoare de TME și EGFR la animalele gestante a determinat o incidență crescută a afectării dezvoltării embrio-fetale, a mortalității embrionare și a avortului. Prin urmare, pe baza mecanismului său de acțiune și a rezultatelor obținute pe modele animale, amivantamab poate dăuna fătului atunci când este administrat unei femei gravide. Amivantamabul nu trebuie administrat în timpul sarcinii, cu excepția cazului în care se consideră că beneficiul tratamentului cu amivantamab depășește riscurile potențiale pentru făt. Dacă pacienta rămâne gravidă în timpul tratamentului cu acest medicament, pacienta trebuie informată cu privire la riscul potențial pentru făt (vezi pct. 5.3).
Alăptarea
Nu se cunoaște dacă amivantamabul este excretat în laptele matern uman. Se știe că IgG uman este excretat în laptele matern în primele câteva zile după naștere, concentrația acestuia scăzând ulterior până la valori joase. În timpul acestei scurte perioade nu poate fi exclus un anumit risc pentru copilul alăptat la sân, deși imunoglubulinele de tip IgG sunt degradate în tractul gastro-intestinal al sugarului alăptat la sân și, astfel, nu sunt absorbite. Trebuie luată o decizie fie de a întrerupe alăptarea, fie de a întrerupe/a omite tratamentul cu amivantamab, având în vedere beneficiul alăptării pentru copil și beneficiul tratamentului pentru femeie.
Fertilitatea
Nu sunt disponibile date pentru determinarea efectelor amivantamabului asupra fertilității la om. Efectele asupra fertilității la bărbați și femei nu au fost evaluate în cadrul unor studii efectuate pe animale.
4.7 Efecte asupra capacității de a conduce vehicule și de a folosi utilaje
Rybrevant poate avea o influență moderată asupra capacității de a conduce vehicule și de a folosi utilaje. Vă rugăm să consultați pct. 4.8 (de ex., amețeală, fatigabilitate, tulburări de vedere). Dacă în urma tratamentului pacienții prezintă simptome, inclusiv reacții adverse legate de vedere, care le afectează capacitatea de concentrare și reacție, acestora li se recomandă să nu conducă sau să nu folosească utilaje până la dispariția efectului medicamentului.
4.8 Reacții adverse
Rezumatul profilului de siguranță
În setul de date referitoare la amivantanab administrat în monoterapie (N=380), cele mai frecvente reacții adverse de toate gradele au fost erupții cutanate tranzitorii (76%), reacții adverse legate de perfuzie (67%), toxicitate unghială (47%), hipoalbuminemie (31%), edem (26%), fatigabilitate (26%), stomatită (24%), greață (23%) și constipație (23%). Reacțiile adverse grave au inclus BPI (1,3%), RALP (1,1%) și erupții cutanate (1,1%). Trei procente dintre pacienți au întrerupt tratamentul cu
Rybrevant din cauza reacțiilor adverse. Cele mai frecvente reacții adverse care au dus la întreruperea tratamentului au fost RALP (1,1%), BPI (0,5%) și toxicitatea unghială (0,5%).
Tabelul reacțiilor adverse
Tabelul 7 prezintă pe scurt reacțiile adverse la medicament care au apărut la pacienții cărora li s-a administrat amivantamab în monoterapie.
Datele reflectă expunerea la amivantamab a 380 de pacienți cu cancer pulmonar fără celule mici, avansat local sau metastazat, după eșecul chimioterapiei pe bază de platină. Pacienților li s-a administrat amivantamab 1050 mg (pentru pacienții cu greutatea < 80 kg) sau 1400 mg (pentru pacienții cu greutatea ≥80 kg). Valoarea mediană a expunerii la amivantamab a fost de 4,1 luni (interval: 0,0 până la 39,7 luni).
Reacțiile adverse observate în timpul studiilor clinice sunt enumerate mai jos pe categorii de frecvență. Categoriile de frecvențe sunt definite după cum urmează: foarte frecvente (≥1/10); frecvente (≥1/100 și < 1/10); mai puțin frecvente (≥1/1000 și < 1/100); rare (≥1/10000 și < 1/1000); foarte rare (< 1/10000); cu frecvență necunoscută (care nu poate fi estimată din datele disponibile).
În fiecare categorie de frecvență, reacțiile adverse sunt prezentate în ordinea descrescătoare a gravității.
Tabelul 7: Reacții adverse la pacienții cărora li se administrează amivantamab în monoterapie
| Sisteme şi organe Reacții adverse | Categoria de frecvenţă | Toate gradele (%) | Grad 3-4 (%) |
|---|---|---|---|
| Tulburări metabolice și de nutriție | |||
| Hipoalbuminemie*(vezi pct.5.1) | Foarte frecvente | 31 | 2† |
| Scăderea poftei de mâncare | 16 | 0,5† | |
| Hipocalcemie | 10 | 0,3† | |
| Hipopotasemie | Frecvente | 9 | 2 |
| Hipomagnezemie | 8 | 0 | |
| Tulburări ale sistemului nervos | |||
| Amețeală* | Foarte frecvente | 13 | 0,3† |
| Tulburări oculare | |||
| Tulburări de vedere* | Frecvente | 3 | 0 |
| Creșterea genelor* | 1 | 0 | |
| Alte tulburări oculare* | 6 | 0 | |
| Cheratită | Mai puțin frecvente | 0,5 | 0 |
| Uveită | 0,3 | 0 | |
| Tulburări respiratorii, toracice și mediastinale | |||
| Boală pulmonară interstițială* | Frecvente | 3 | 0,5† |
| Tulburări gastro-intestinale | |||
| Diaree | Foarte frecvente | 11 | 2† |
| Stomatită* | 24 | 0,5† | |
| Greață | 23 | 0,5† | |
| Constipație | 23 | 0 | |
| Vărsături | 12 | 0,5† | |
| Durere abdominală* | Frecvente | 9 | 0,8† |
| Hemoroizi | 3,7 | 0 | |
| Tulburări hepatobiliare | |||
| Valori crescute ale alanin-aminotransferazei | Foarte frecvente | 15 | 2 |
| Valori crescute ale aspartat- aminotransferazei | 13 | 1 | |
| Valori crescute ale fosfatazei alcaline serice | 12 | 0,5† | |
| Afecțiuni cutanate și ale țesutului subcutanat | |||
| Erupție cutanată tranzitorie* | Foarte frecvente | 76 | 3† |
| Toxicitate unghială* | 47 | 2† | |
| Xerodermie* | 19 | 0 | |
| Prurit | 18 | 0 | |
| Ulcerație cutanată | Mai puțin frecvente | 0,8 | 0 |
| Necroliză epidermică toxică | 0,3 | 0,3† | |
| Tulburări musculo-scheletice și ale țesutului conjunctiv | |||
| Mialgie | Foarte frecvente | 11 | 0,3† |
| Tulburări generale și la nivelul locului de administrare | |||
| Edem* | Foarte frecvente | 26 | 0,8† |
| Fatigabilitate* | 26 | 0,8† | |
| Pirexie | 11 | 0 | |
| Leziuni, intoxicații și complicații legate de procedurile utilizate | |||
| Reacții adverse legate de perfuzie | Foarte frecvente | 67 | 2 |
* Termeni grupați
† Doar reacții de Grad 3
Rezumatul profilului de siguranță
În setul de date referitoare la amivantamab administrat în asociere cu carboplatină și pemetrexed (N=301), cele mai frecvente reacții adverse de toate gradele au fost erupții cutanate (83%), neutropenie (57%), toxicitate unghială (53%), reacții adverse legate de perfuzie (51%), fatigabilitate (43%), stomatită (39%), greață (43%), trombocitopenie (40%), constipație (40%), edem (40%), scăderea poftei de mâncare (33%), hipoalbuminemie (32%), valori crescute ale alaninaminotransferazei (26%), valori crescute ale aspartat-aminotransferazei (23%), vărsături (22%) și hipopotasemie (20%). Reacțiile adverse grave au inclus erupții cutanate (2,7%), tromboembolism venos (2,3%), trombocitopenie (2,3%) și BPI (2,0%). Opt la sută dintre pacienți au întrerupt tratamentul cu Rybrevant din cauza reacțiilor adverse. Cele mai frecvente reacții adverse care au condus la întreruperea tratamentului au fost RALP (2,7%), erupții cutanate (2,3%), BPI (2,3%), și toxicitate unghială (1,0%).
Tabelul 8 prezintă pe scurt reacțiile adverse la medicamente care au apărut la pacienții cărora li s-a administrat amivantamab în asociere cu chimioterapia.
Datele reflectă expunerea la amivantamab în asociere cu carboplatină și pemetrexed la 301 pacienți cu cancer pulmonar cu celule mici, avansat local sau metastazat. Pacienților li s-a administrat amivantamab 1400 mg (pentru pacienți < 80 kg) sau 1750 mg (pentru pacienți ≥ 80 kg) săptămânal, timp de 4 săptămâni. Începând cu săptămâna 7, pacienților li s-a administrat amivantamab 1750 mg (pentru pacienți < 80 kg) sau 2100 mg (pentru pacienți ≥ 80 kg) la fiecare 3 săptămâni. Expunerea medie la amivantamab în asociere cu carboplatină și pemetrexed a fost de 7,7 luni (interval: 0,0 până la 28,1 luni).
Reacțiile adverse observate în timpul studiilor clinice sunt enumerate mai jos în funcție de categoria de frecvență. Categoriile de frecvență sunt definite după cum urmează: Foarte frecvente (≥1/10); frecvente (≥1/100 și < 1/10); mai puțin frecvente (≥1/1000 și < 1/100); rare (≥1/10000 și < 1/1000); foarte rare (< 1/10000); și cu frecvență necunoscută (care nu poate fi estimată din datele disponibile).
În cadrul fiecărei grupe de frecvență, reacțiile adverse sunt prezentate în ordinea descrescătoare a gravității.
Tabelul 8: Reacții adverse la pacienții carora li se administrează amivantamab în asociere cu carboplatină și pemetrexed
| Sisteme și organe Reacții adverse | Categoria de frecvență | Toate gradele (%) | Grad 3-4 (%) |
|---|---|---|---|
| Tulburări hematologice și limfatice | |||
| Neutropenie | Foarte frecvente | 57 | 39 |
| Trombocitopenie | 40 | 12 | |
| Tulburări metabolice și de nutriție | |||
| Scăderea poftei de mâncare | Foarte frecvente | 33 | 1,3 |
| Hipoalbuminemie* | 32 | 3,7 | |
| Hipopotasemie | 20 | 6,6 | |
| Hipomagnezemie | 13 | 1,3 | |
| Hipocalcemie | 12 | 1,0 | |
| Tulburări ale sistemului nervos | |||
| Amețeală* | Frecvente | 10 | 0,3 |
| Tulburări vasculare | |||
| Tromboembolism venos* | Foarte frecvente | 14 | 3,0 |
| Tulburări oculare | |||
| Alte tulburări oculare* | Frecvente | 7,3 | 0 |
| Tulburări de vedere* | 3,0 | 0 | |
| Creșterea genelor | Mai puțin frecvente | 0,3 | 0 |
| Cheratită | 0,3 | 0 | |
| Uveită | 0,3 | 0 | |
| Tulburări respiratorii, toracice și mediastinale | |||
| Boală pulmonară interstițială* | Frecvente | 2,3 | 1,7 |
| Tulburări gastro-intestinale | |||
| Greață | Foarte frecvente | 43 | 1,0 |
| Constipație | 40 | 0,3 | |
| Stomatită* | 39 | 3,0 | |
| Vărsături | 22 | 2,0 | |
| Diaree | 19 | 2,3 | |
| Durere abdominală* | Frecvente | 11 | 0,3 |
| Hemoroizi | 9,3 | 0,7 | |
| Tulburări hepatobiliare | |||
| Valori crescute ale alanin- aminotransferazei | Foarte frecvente | 26 | 4,3 |
| Valori crescute ale aspartat- aminotransferazei | 23 | 0,7 | |
| Valori crescute ale fosfatazei alcaline serice | Frecvente | 10 | 0,3 |
| Afecțiuni cutanate și ale țesutului subcutanat | |||
| Erupție cutanată* | Foarte frecvente | 83 | 14 |
| Toxicitate unghială* | 53 | 4,3 | |
| Xerodermie* | 16 | 0 | |
| Prurit | 10 | 0 | |
| Ulcerație cutanată | Frecvente | 3,7 | 0,7 |
| Tulburări musculo-scheletice și ale țesutului conjunctiv | |||
| Mialgie | Frecvente | 5,0 | 0,7 |
| Tulburări generale și la nivelul locului de administrare | |||
| Fatigabilitate* | Foarte frecvente | 43 | 4,7 |
| Edem* | 40 | 1,3 | |
| Pirexie | 14 | 0 | |
| Leziuni, intoxicații și complicații legate de procedurile utilizate | |||
| Reacții adverse legate de perfuzie | Foarte frecvente | 51 | 3,0 |
* Termeni grupați
Rezumatul profilului de siguranță
În setul de date referitor la amivantamab în asociere cu lazertinib (N=421), cele mai frecvente reacții adverse de toate gradele au fost erupții cutanate tranzitorii (89%), toxicitate la nivelul unghiilor (71%), reacții adverse legate de perfuzie (63%), hipoalbuminemie (48%), hepatotoxicitate (47%), edem (47%), stomatită (43%), tromboembolism venos (37%), parestezie (lazertinib) (34%), fatigabilitate (32%), diaree (29%), constipație (29%), xerodermie (26%), prurit (24%), scăderea poftei de mâncare (24%), hipocalcemie (21%), greață (21%) și alte tulburări oculare (21%). Cele mai frecvente reacții adverse grave au inclus tromboembolism venos (11%), penumonie (4,0%), erupție cutanată tranzitorie (3,1%), BPI/pneumonită (2,9%), hepatotoxicitate (2,4%), COVID-19 (2,4%) și RALP și efuziune pleurală (2,1%). 23% dintre pacienți au întrerupt tratamentul cu Rybrevant din cauza reacțiilor adverse. Cele mai frecvente reacții adverse care au dus la întreruperea tratamentului cu Rybrevant au fost erupție cutanată tranzitorie (5,5%), reacții adverse legate de perfuzie (4,5%), toxicitate unghială (3,6%), BPI (2,9%) și TEV (2,9%).
Tabelul 9 prezintă pe scurt reacțiile adverse la medicament care au apărut la pacienții cărora li s-a administrat amivantamab în asociere cu lazertinib.
Datele reflectă expunerea la amivantamab în asociere cu lazertinib la 421 de pacienți cu cancer pulmonar fără celule mici avansat local sau metastazat. Pacienților li s-a administrat amivantamab 1050 mg (la pacienți cu greutatea < 80 kg) sau 1400 mg (la pacienți cu greutatea ≥80 kg) o dată pe săptămână, timp de 4 săptămâni, apoi o dată la 2 săptămâni. Expunerea mediană la tratamentul de studiu în grupul de tratament cu amivantamab în asociere cu lazertinib a fost de 18,5 luni (interval: 0,2 până la 31,4 luni).
Reacțiile adverse observate în timpul studiilor clinice sunt enumerate mai jos în funcție de frecvență. Frecvențele sunt definite după cum urmează: foarte frecvente (≥1/10), frecvente (≥1/100 și < 1/10), mai puțin frecvente (≥1/1 000 și < 1/100), rare (≥1/10 000 și < 1/1 000), foarte rare (< 1/10 000) și cu frecvență necunoscută (care nu poate fi estimată din datele disponibile).
În cadrul fiecărei grupe de frecvență, reacțiile adverse sunt prezentate în ordinea descrescătoare a gravității.
Tabelul 9: Reacții adverse la pacienții tratați cu amivantamab în asociere cu lazertinib
| Clasa de aparate, sisteme și organe Reacție adversă | Categoria de frecvență | Orice grad (%) | Grad 3-4 (%) |
|---|---|---|---|
| Tulburări metabolice și de nutriție | |||
| Hipoalbuminemie* | Foarte frecvente | 48 | 5 |
| Apetit alimentar scăzut | 24 | 1,0 | |
| Hipocalcemie | 21 | 2,1 | |
| Hipopotasemie | 14 | 3,1 | |
| Hipomagneziemie | Frecvente | 5,0 | 0 |
| Tulburări ale sistemului nervos | |||
| Paraestezie*‡ | Foarte frecvente | 34 | 1,7 |
| Amețeală* | 13 | 0 | |
| Tulburări vasculare | |||
| Tromboembolism venos* | Foarte frecvente | 37 | 11 |
| Tulburări oculare | |||
| Alte tulburări oculare* | Foarte frecvente | 21 | 0,5 |
| Tulburări de vedere* | Frecvente | 4,5 | 0 |
| Cheratită | 2,6 | 0,5 | |
| Creșterea genelor* | 1,9 | 0 | |
| Tulburări respiratorii, toracice și mediastinale | |||
| Boală pulmonară interstițială/pneumonită* | Frecvente | 3,1 | 1,2 |
| Tulburări gastro-intestinale | |||
| Stomatită* | Foarte frecvente | 43 | 2,4 |
| Diaree | 29 | 2,1 | |
| Constipație | 29 | 0 | |
| Greață | 21 | 1,2 | |
| Vărsături | 12 | 0,5 | |
| Dureri abdominale* | 11 | 0 | |
| Hemoroizi | Frecvente | 10 | 0,2 |
| Tulburări hepatobiliare | |||
| Hepatotoxicitate† | Foarte frecvente | 47 | 9 |
| Afecțiuni cutanate și ale țesutului subcutanat | |||
| Erupție cutanată tranzitorie* | Foarte frecvente | 89 | 27 |
| Toxicitate la nivelul unghiilor* | 71 | 11 | |
| Piele uscată* | 26 | 1,0 | |
| Prurit | 24 | 0,5 | |
| Sindromul de eritrodisestezie palmo- plantară | Frecvente | 6 | 0,2 |
| Ulcerație cutanată | 5 | 0,7 | |
| Urticarie | 1,2 | 0 | |
| Tulburări musculo-scheletice și ale țesutului conjunctiv | |||
| Spasme musculare | Foarte frecvente | 17 | 0,5 |
| Mialgie | 13 | 0,7 | |
| Tulburări generale și la nivelul locului de administrare | |||
| Edem* | Foarte frecvente | 47 | 2,9 |
| Fatigabilitate* | 32 | 3,8 | |
| Pirexie | 12 | 0 | |
| Leziuni, intoxicații și complicații legate de proceduri | |||
| Reacție adversă legată de perfuzie | Foarte frecvente | 63 | 6 |
* Termeni grupați
‡
Evaluată ca reacție adversă numai pentru lazertinib.
†
Cele mai frecvente evenimente au inclus creșterea valorilor ALT (36%), creșterea valorilor AST (29%) și creșterea fosfatazei alcaline sanguine (12%).
Descrierea reacțiilor adverse selectate
Reacții adverse legate de perfuzie
La pacienții tratați cu amivantamab în monoterapie, reacțiile adverse legate de perfuzie au apărut la 67% dintre pacienți. 98% din RALP-uri au fost de gradul 1-2. 99% din RALP-uri au apărut la prima perfuzie, cu un timp median până la debut de 60 de minute, iar majoritatea au survenit în primele 2 ore de la debutul perfuziei. Cele mai frecvente semne și simptome includ frisoane, dispnee, greață, înroșirea feței, disconfort toracic și vărsături (vezi pct. 4.4).
Reacțiile adverse legate de perfuzie au apărut la 50% dintre pacienții tratați cu amivantamab în asociere cu carboplatină și pemetrexed. Peste 94% din RALP-uri au fost de gradul 1-2. Majoritatea RALP-urilor au apărut la prima perfuzie, cu un timp median până la debut de 60 de minute (interval de 0-7 ore), iar majoritatea au survenit în primele 2 ore de la debutul perfuziei.
Ocazional, RALP-urile pot apărea la reluarea administrării de amivantamab după o perioadă prelungită de întrerupere a administrării dozei de peste 6 săptămâni.
La pacienții tratați cu amivantamab în asociere cu lazertinib, reacțiile adverse legate de perfuzie au apărut la 63% dintre pacienți. 94% dintre RALP-uri au fost de grad 1-2. Majoritatea RALP-urilor au apărut la prima perfuzie, cu un timp median până la debut de 1 oră, iar majoritatea au apărut în decurs de 2 ore de la începerea perfuziei. Cele mai frecvente semne și simptome includ frisoane, dispnee, greață, înroșirea feței, disconfort toracic și vărsături (vezi pct. 4.4).
Ocazional, poate să apară o RALP la reluarea tratamentului cu amivantamab după întreruperi prelungite ale dozei de peste 6 săptămâni.
Într-un studiu de fază 2, deschis, multicentric, efectuat la pacienții cu NSCLC, pacienților li s-au administrat 8 mg dexametazonă pe cale orală, de două ori pe zi, în fiecare din cele două zile înainte de prima perfuzie cu Rybrevant și 8 mg administrat oral, cu 60 de minute înainte de perfuzie în ziua primei perfuzii (5 doze în total), suplimentar la perfuzia cu dexametazonă. Odată cu adăugarea de dexametazonă administrată oral, în ziua perfuziei inițiale s-a raportat o incidență de 22,5% a RALP și nicio RALP de grad ≥3 (vezi pct. 4.2).
Boală pulmonară interstițială
Boala pulmonară interstițială sau reacțiile adverse similare BPI au fost raportate la utilizarea amivantamab, precum și la administrarea altor inhibitori ai EGFR. Boala pulmonară interstițială sau pneumonita a fost raportată la 2,6% dintre pacienții tratați cu amivantamab în monoterapie, la 2,3% dintre pacienții tratați cu amivantamab în asociere cu carboplatină și pemetrexed și la 3,1% dintre pacienții tratați cu amivantamab în asociere cu lazertinib, inclusiv 1 (0,2%) caz mortal. Pacienții cu istoric medical de BPI, BPI indusă medicamentos, pneumonită de iradiere care a necesitat tratament cu steroizi sau orice dovadă de BPI activă clinic au fost excluși din studiul clinic (vezi pct. 4.4).
Evenimente tromboembolice venoase (TEV) asociate cu administrarea în asociere cu lazertinib
Atunci când Rybrevant este utilizat în asociere cu lazertinib, evenimentele TEV, inclusiv tromboza venoasă profundă (TVP) și embolia pulmonară (EP), au fost raportate la 37% dintre cei 421 de pacienți cărora li s-a administrat Rybrevant în asociere cu lazertinib. Majoritatea evenimentelor au fost de gradul 1 sau 2, evenimentele de gradul 3-4 survenind la 11% dintre pacienții cărora li s-a administrat Rybrevant în asociere cu lazertinib, iar decesele survenind la 0,5% dintre pacienții cărora li s-a administrat Rybrevant în asociere cu lazertinib. Pentru informații privind anticoagulantele profilactice și abordarea terapeutică a evenimentelor TEV, vezi pct. 4.2 și 4.4.
La pacienții tratați cu Rybrevant în asociere cu lazertinib, mediana intervalului de timp până la primul debut al evenimentului TEV a fost de 84 zile. Evenimentele TEV au dus la oprirea tratamentului cu Rybrevant la 2,9% dintre pacienți.
Reacții cutanate și unghiale
Erupțiile cutanate tranzitorii (inclusiv dermatita acneiformă), pruritul și pielea uscată au apărut la 76% dintre pacienții tratați cu amivantamab în monoterapie. Majoritatea cazurilor au fost de grad 1 sau 2, cu evenimente de erupții cutanate tranzitorii de gradul 3 care au apărut la 3% dintre pacienți. Erupțiile cutanate tranzitorii care au dus la întreruperea tratamentului cu amivantamab au apărut la 0,3% dintre pacienți. Erupțiile cutanate au apărut, de obicei, în primele 4 săptămâni de tratament, cu un timp median până la debut de 14 zile. Au fost înregistrate cazuri de toxicitate unghială la pacienții tratați cu amivantamab. Majoritatea evenimentelor au fost de grad 1 sau 2, toxicitatea unghială de grad 3 apărând la 1,8% dintre pacienți.
Erupțiile cutanate tranzitorii (inclusiv dermatita acneiformă) au apărut la 83% dintre pacienții tratați cu amivantamab în asociere cu carboplatină și pemetrexed. Majoritatea cazurilor au fost de grad 1 sau 2, cu evenimente de erupții cutanate tranzitorii de gradul 3 apărând la 14% dintre pacienți. Erupțiile cutanate tranzitorii care au dus la întreruperea tratamentului cu amivantamab au apărut la 2,3% dintre pacienți. Erupțiile cutanate au apărut, de obicei, în primele 4 săptămâni de tratament, cu un timp median până la debut de 14 zile. Au fost înregistrate cazuri de toxicitate unghială la pacienții tratați cu amivantamab în asociere cu carboplatină și pemetrexed. Majoritatea evenimentelor au fost de grad 1 sau 2, toxicitatea unghială de grad 3 apărând la 4,3 % dintre pacienți (vezi pct. 4.4).
Erupțiile cutanate tranzitorii (inclusiv dermatita acneiformă) au apărut la 89% dintre pacienții tratați cu amivantamab în asociere cu lazertinib. Majoritatea cazurilor au fost de grad 1 sau 2, cu evenimente de erupții cutanate tranzitorii de gradul 3 care au apărut la 27% dintre pacienți. Erupțiile cutanate tranzitorii care au dus la întreruperea administrării de amivantamab au apărut la 5,5% dintre pacienți. Erupțiile cutanate tranzitorii au apărut de regulă în primele 4 săptămâni de tratament, cu o mediană a intervalului de timp până la debut de 14 zile. La pacienții tratați cu amivantamab în asociere cu lazertinib a apărut toxicitatea la nivelul unghiilor. Majoritatea evenimentelor au fost de grad 1 sau 2, evenimentele de toxicitate la nivelul unghiilor de gradul 3 survenind la 11% dintre pacienți (vezi pct. 4.4).
A fost realizat un studiu de fază 2 la pacienții tratați cu Rybrevant în asociere cu lazertinib pentru a evalua utilizarea terapiei profilactice cu un antibiotic cu utilizare orală, un antibiotic administrat topic la nivelul scalpului, o cremă hidratantă pe față și pe întreg corpul (cu excepția scalpului) și un antiseptic pe mâini și picioare (vezi pct. 4.2 și 4.4). S-a demonstrat o reducere a incidenței evenimentelor adverse dermatologice de grad ≥2 în primele 12 săptămâni de tratament, comparativ cu tratamentul dermatologic standard utilizat în practica clinică (38,6% față de 76,5%, p<0,0001). În plus, s-a observat o reducere a evenimentelor adverse de grad ≥2 care au afectat scalpul în primele 12 săptămâni de tratament (8,6% față de 29,4%), împreună cu o incidență mai mică a reducerilor de doză (7,1% față de 19,1%), a întreruperilor administrării (15,7% față de 33,8%) și a opririi tratamentului (1,4% față de 4,4%) din cauza evenimentelor adverse dermatologice.
Tulburări oculare
Tulburările oculare, inclusiv cheratita (0,5%), au apărut la 9% dintre pacienții tratați cu amivantamab în monoterapie. Alte reacții adverse raportate au inclus creșterea genelor, afectarea vederii și alte tulburări oculare. Toate evenimentele au fost de grad 1-2.
Tulburările oculare, inclusiv cheratita (0,3%), au apărut la 11% dintre pacienții tratați cu amivantamab în asociere cu carboplatină și pemetrexed. Alte reacții adverse raportate au inclus creșterea genelor, afectarea vederii, uveită și alte tulburări oculare. Toate evenimentele au fost de grad 1-2 (vezi pct. 4.4).
La pacienții tratați cu amivantamab în asociere cu lazertinib au apărut tulburări oculare, inclusiv cheratită (2,6%). Alte reacții adverse raportate au inclus creștere a genelor, tulburări de vedere și alte tulburări oculare. Majoritatea evenimentelor au fost de grad 1-2 (vezi pct. 4.4).
Categorii speciale de populație
Vârstnici
Există informații clinice limitate privind administrarea amivantamab la pacienții cu vârstă ≥ 75 ani (vezi pct. 5.1). În general, nu s-au observat diferențe în ceea ce privește siguranța administrării la pacienții cu vârstă ≥ 65 ani față de pacienții cu vârstă < 65 ani.
Imunogenitatea
Similar tuturor proteinelor terapeutice, există un potențial de imunogenitate. În studiile clinice efectuate la pacienți cu NSCLC avansat local sau metastazat tratați cu amivantamab, 4 (0,2%) dintre cei 1862 de participanți cărora li s-a administrat Rybrevant și care au fost evaluabili pentru prezența anticorpilor anti-medicament (AAM), au fost testați pozitiv pentru anticorpii anti-amivantamab apăruți ca urmare a tratamentului. Nu au existat dovezi de modificare a profilului farmacocinetic, a eficacității sau a profilului de siguranță din cauza apariției anticorpilor anti-amivantamab.
Raportarea reacțiilor adverse suspectate
Raportarea reacțiilor adverse suspectate după autorizarea medicamentului este importantă. Acest lucru permite monitorizarea continuă a raportului beneficiu/risc al medicamentului. Profesioniștii din domeniul sănătății sunt rugați să raporteze orice suspiciune de reacție adversă prin intermediul sistemului național de raportare, așa cum este menționat în Anexa V.
4.9 Supradozaj
Nu s-a determinat doza maximă tolerată într-un studiu clinic în care pacienților li s-au administrat intravenos până la 2100 mg. Nu se cunoaște un antidot specific pentru supradozajul cu amivantamab. În cazul unui supradozaj, tratamentul cu Rybrevant trebuie întrerupt, pacientul trebuie monitorizat pentru orice semne sau simptome de evenimente adverse și trebuie instituite imediat măsuri generale adecvate de asistență până la diminuarea sau remisiunea toxicității clinice.
5. PROPRIETĂȚI FARMACOLOGICE
5.1 Proprietăți farmacodinamice
Grupa farmacoterapeutică: Anticorpi monoclonali și conjugați anticorp-medicament, codul ATC: L01FX18.
Mecanism de acțiune
Amivantamab este un anticorp bispecific EGFR-TME cu conținut scăzut de fucoză, complet uman, de tip IgG1, cu activitate imunitară mediată celular, care vizează tumorile cu mutații activatoare de EGFR, cum ar fi delețiile în Exonul 19, substituția în Exonul 21 L858R și mutațiile de inserție în Exonul 20. Amivantamab se leagă de domeniile extracelulare ale EGFR și TME.
Amivantamab întrerupe funcțiile de semnalizare ale EGFR și TME prin blocarea legării ligandului și intensificarea degradării EGFR și a TME, prevenind astfel creșterea și progresia tumorii. Prezența EGFR și MET pe suprafața celulelor tumorale permite, de asemenea, țintirea acestor celule pentru distrugere de către celulele efectoare imune, cum ar fi celulele natural killer și macrofagele, prin citotoxicitate dependentă de anticorpi mediată celular (CDAC) și, respectiv, mecanisme de trogocitoză.
Efecte farmacodinamice
Albumină
Amivantamabul a scăzut concentrația serică de albumină, un efect farmacodinamic al inhibării TME, de obicei în primele 8 săptămâni (vezi pct. 4.8); ulterior, concentrația de albumină s-a stabilizat pentru restul tratamentului cu amivantamab.
Eficacitate și siguranță clinică
Cancer pulmonar fără celule mici (NSCLC) netratat anterior, cu deleții în Exonul 19 al EGFR sau cu mutații de substituție în Exonul 21 L858R (MARIPOSA)
NSC3003 (MARIPOSA) este un studiu multicentric de fază 3, randomizat, în regim deschis, controlat activ, de evaluare a eficacității și a siguranței Rybrevant în asociere cu lazertinib comparativ cu osimertinib în monoterapie ca tratament de primă linie la pacienții cu NSCLC cu mutații EGFR, avansat local sau metastatic, care nu răspunde la terapia curativă. Trebuia ca probele pacienților să aibă una dintre cele două mutații comune din EGFR (deleție a Exonului 19 sau mutație de substituție L858R a Exonului 21), identificată prin testare la nivel local. Probele de țesut tumoral (94%) și/sau de plasmă (6%) prelevate de la toți pacienții au fost testate la nivel local pentru a determina statusul deleției Exonului 19 al EGFR și/sau al mutației de substituție L858R a Exonului 21, utilizând reacția de polimerizare în lanț (polymerase chain reaction, PCR) la 65% dintre pacienți și secvențierea de generație următoare (NGS) la 35% dintre pacienți.
În total, 1074 de pacienți au fost randomizați (în raport de 2:2:1) pentru a li se administra Rybrevant în asociere cu lazertinib, osimertinib în monoterapie sau lazertinib în monoterapie până la progresia bolii sau până la toxicitate inacceptabilă. Rybrevant a fost administrat intravenos în doze de 1050 mg (la pacienți cu greutatea < 80 kg) sau de 1400 mg (la pacienți cu greutatea ≥80 kg), o dată pe săptămână timp de 4 săptămâni, și ulterior, începând cu săptămâna 5, la fiecare 2 săptămâni. Lazertinib a fost administrat în doze de 240 mg oral o dată pe zi. Osimertinib a fost administrat în doze de 80 mg oral o dată pe zi. Randomizarea a fost stratificată în funcție de tipul mutației din EGFR (deleție a Exonului 19 sau Exonul 21 L858R), rasă (asiatică sau non-asiatică) și antecedente de metastaze cerebrale (da sau nu).
Caracteristicile demografice și ale bolii la momentul inițial au fost echilibrate între grupurile de tratament. Mediana vârstei a fost de 63 (interval: 25-88) de ani, 45% dintre pacienți având vârsta ≥65 de ani; 62% au fost femei; 59% au fost asiatici și 38% caucazieni. Statusul de performanță la momentul inițial al Grupului Estic pentru Cooperare în Oncologie (Eastern Cooperative Oncology Group, ECOG) a fost 0 (34%) sau 1 (66%); 69% nu fumaseră niciodată; 41% aveau metastaze cerebrale anterioare, iar 90% aveau cancer în stadiul IV la diagnosticul inițial. În ceea ce privește statusul mutațiilor la nivelul EGFR, 60% erau deleții ale Exonului 19 și 40% erau mutații de substituție L858R ale Exonului 21.
Rybrevant în asociere cu lazertinib a demonstrat o îmbunătățire semnificativă statistic a supraviețuirii fără progresia bolii (SFP) pe baza evaluării BICR.
Analiza finală privind SG a demonstrat o îmbunătățire semnificativă din punct de vedere statistic a SG în cazul tratamentului cu Rybrevant în asociere cu lazertinib în comparație cu tratamentul cu osimertinib (vezi Tabelul 10 și Figura 2).
Tabelul 10: Rezultate privind eficacitatea în studiul MARIPOSA
| Rybrevant + lazertinib (N=429) | Osimertinib (N=429) | |
| Supraviețuire fără progresia bolii (SFP)a | ||
|---|---|---|
| Număr de evenimente | 192 (45%) | 252 (59%) |
| Mediană, luni (IÎ 95%) | 2,7 (19,1, 27,7) | 16,6 (14,8, 18,5) |
| Risc relativ (IÎ 95%); valoarea p | 0,70 (0,58, 0,85); p=0,0002 | |
| Supraviețuire globală (SG) | ||
| Număr de evenimente | 173 (40%) | 217 (51%) |
| Mediană, luni (IÎ 95%) | NE (42,9, NE) | 36,7 (33,4, 41,0) |
| Risc relativ (IÎ 95%); valoarea p | 0,75 (0,61, 0,92); p=0,0048 | |
| Rata răspunsului obiectiv (RRO)a, b | ||
| RRO % (IÎ 95%) | 80% (76%, 84%) | 77% (72%, 81%) |
| Durata răspunsului (DR)a,b | ||
| Mediană, luni (IÎ 95%) | 25,8 (20,3, 33,9) | 18,1 (14,8, 20,1) |
BICR = analiză centrală independentă în regim orb; IÎ = interval de încredere; NE = nu se poate estima.
Rezultatele privind SFP se referă la data centralizării datelor 11 august 2023, cu o perioadă mediană de urmărire de 22,0 luni. Rezultatele privind RRO și DR se referă la data centralizării datelor 13 mai 2024, cu o perioadă mediană de urmărire de 31,3 luni. Rezultatele privind SG se referă la data centralizării datelor 4 decembrie 2024, cu o perioadă mediană de urmărire de 37,8 luni.
a
BICR pe baza criteriilor RECIST v1.1. b
Pe baza respondenților confirmați.
Figura 1: Curba Kaplan-Meier a SFP la pacienți cu NSCLC netratați anterior, obținută în urma evaluării BICR
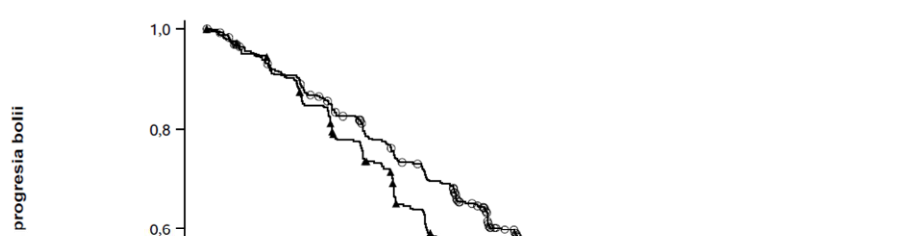


Figura 2: Curba Kaplan-Meier a SG la pacienți cu NSCLC netratați anterior



În studiul MARIPOSA, RRO intracranian și DR pe baza evaluării BICR au fost criterii finale de evaluare pre-specificate. În subsetul de pacienți cu leziuni intracraniene la momentul inițial, asocierea Rybrevant și lazertinib a demonstrat un RRO intracranian similar cu cel al elementului de control. Conform protocolului, toți pacienții din studiul MARIPOSA au fost supuși unei serii de examinări IRM pentru a evalua răspunsul intracranian și durata. Rezultatele sunt prezentate pe scurt în Tabelul 11.
Tabelul 11: RRO intracranian și DR pe baza evaluării BICR la subiecții cu leziuni intracraniene la momentul inițial - MARIPOSA
| Rybrevant + lazertinib (N=180) | Osimertinib (N=186) | |
| Evaluarea răspunsului tumoral intracranian | ||
|---|---|---|
| RRO intracranian (RC+RP), % (IÎ 95%) | 78% (71%, 84%) | 77% (71%, 83%) |
| Răspuns complet | 64% | 59% |
| DR intracranian | ||
| Număr de respondenți | 140 | 144 |
| Mediană, luni (IÎ 95%) | 35,0 (20,4, NE) | 25,1 (22,1, 31,2) |
IÎ = interval de încredere
NE = nu se poate estima
Rezultatele privind RRO intracranian și DR se referă la data centralizării datelor 4 decembrie 2024, cu o perioadă mediană de urmărire de 37,8 luni.
Cancer pulmonar fără celule mici (NSCLC) tratat anterior, cu deleții în Exonul 19 al EGFR sau cu mutații de substituție în Exonul 21 L858R (MARIPOSA-2)
MARIPOSA-2 este un studiu randomizat (2:2:1), în regim deschis, multicentric, de fază 3, efectuat la pacienți cu NSCLC avansat local sau metastazat, cu deleții în Exonul 19 al EGFR sau cu mutații de substituție în Exonul 21 L858R (testarea mutațiilor ar fi putut fi efectuată în momentul sau după diagnosticul de boală local avansată sau metastatică. Testarea nu trebuia repetată la momentul intrării în studiu, odată ce statutul mutației EGFR fusese deja stabilit anterior), după eșecul tratamentului anterior care a inclus un inhibitor de tirozin kinază (TKI) de generația a treia al EGFR. În total, în cadrul studiului au fost randomizați 657 de pacienți, dintre care 263 au primit carboplatină și pemetrexed (CP), iar 131 au primit Rybrevant în asociere cu carboplatină și pemetrexed
(Rybrevant-CP). În plus, 263 de pacienți au fost randomizați pentru a li se administra Rybrevant în asociere cu lazertinib, carboplatină și pemetrexed într-un braț separat al studiului. Rybrevant a fost administrat intravenos în doză de 1400 mg (la pacienți cu greutatea < 80 kg) sau 1750 mg (la pacienți cu greutatea ≥80 kg), o dată pe săptămână, timp de 4 săptămâni, apoi la fiecare 3 săptămâni, în doză de 1750 mg (la pacienți cu greutatea < 80 kg) sau 2100 mg (la pacienți cu greutatea ≥80 kg), începând cu săptămâna 7, până la progresia bolii sau la atingerea unei toxicități inacceptabile. Carboplatina a fost administrată intravenos, la o valoare a ariei de sub curba concentrației plasmatice în funcție de timp de 5 mg/ml pe minut (ASC 5), o dată la 3 săptămâni, timp de până la 12 săptămâni. Pemetrexed a fost administrat intravenos în doză de 500 mg/m2, o dată la 3 săptămâni, până la progresia bolii sau la atingerea unei toxicități inacceptabile.
Pacienții au fost stratificați în funcție de linia de tratament cu osimertinib (prima linie sau a doua linie), de prezența metastazelor cerebrale anterioare (da sau nu) și de apartenența la rasa asiatică (da sau nu).
Pentru cei 394 de pacienți randomizați în brațul Rybrevant-CP sau în brațul CP, vârsta mediană a fost de 62 de ani (interval: 31-85 de ani), cu 38% dintre pacienți având vârsta ≥65 de ani; 60% au fost femei; 48% au fost asiatici și 46% au fost caucazieni. La momentul inițial, scorul de performanță ECOG (Eastern Cooperative Oncology Group) a fost 0 (40%) sau 1 (60%); 66% nu au fumat niciodată; 45% au avut antecedente de metastaze cerebrale și 92% au avut cancer în stadiul IV la diagnosticul inițial.
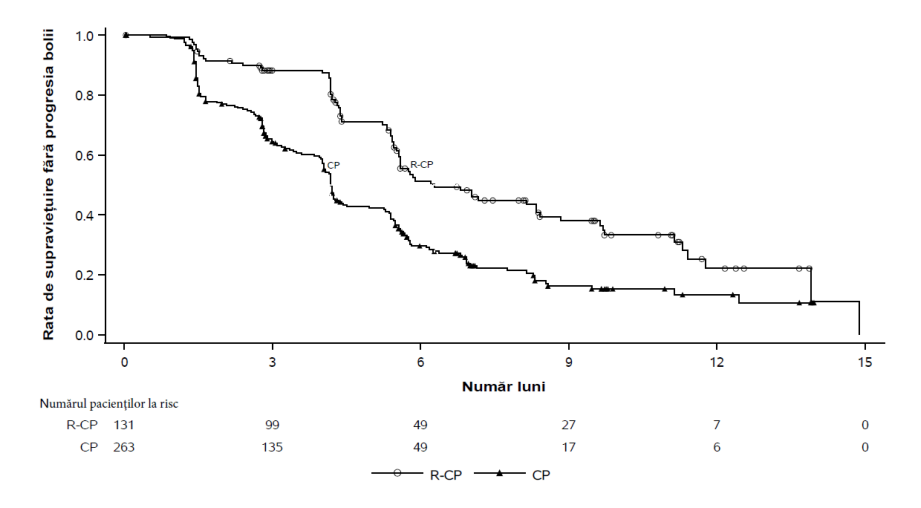
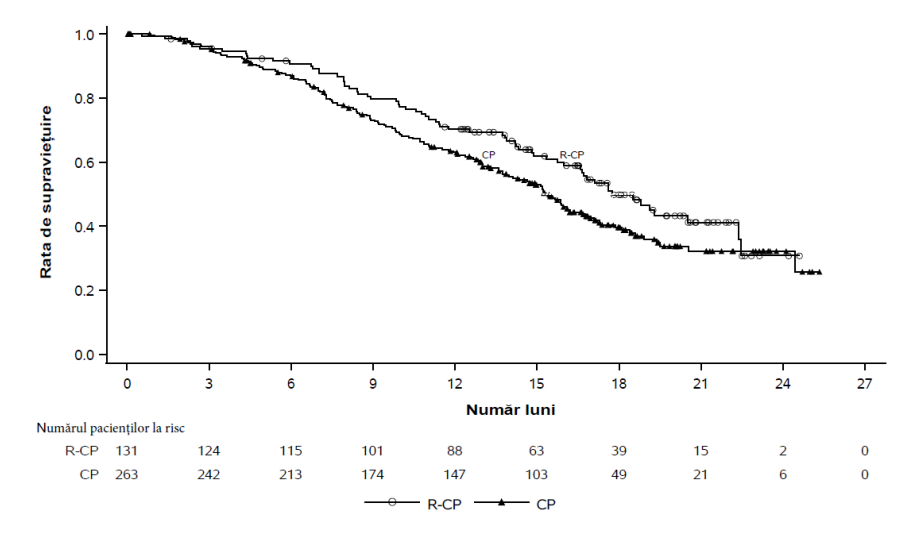
Rybrevant în asociere cu carboplatină și pemetrexed a demonstrat o îmbunătățire semnificativă statistic a supraviețuirii fără progresie a bolii (SFP) comparativ cu carboplatină și pemetrexed, cu un RR de 0,48 (IÎ 95%: 0,36, 0,64; p<0,0001). La momentul celei de-a doua analize intermediare pentru SG, cu o perioadă mediană de urmărire de aproximativ 18,6 luni pentru Rybrevant-CP și aproximativ 17,8 luni pentru CP, RR pentru SG a fost 0,73 (IÎ 95%: 0,54, 0,99; p=0,0386). Acest lucru nu a fost semnificativ statistic (testat la un nivel de semnificație prestabilit de 0,0142).
Rezultatele privind eficacitatea sunt prezentate pe scurt în Tabelul 12.
Tabelul 12: Rezultatele privind eficacitatea în MARIPOSA-2
| Rybrevant+ carboplatină+ pemetrexed (N=131) | carboplatină+ pemetrexed (N=263) | |
| Supraviețuire fără progresie a bolii (SFP)a | ||
|---|---|---|
| Număr de evenimente (%) | 74 (57) | 171 (65) |
| Valoarea mediană, luni (IÎ 95%) | 6,3 (5,6, 8,4) | 4,2 (4,0, 4,4) |
| RR (IÎ 95%); valoarea p | 0,48 (0,36, 0,64); p<0,0001 | |
| Supraviețuirea globală (SG) | ||
| Număr de evenimente (%) | 65 (50) | 143 (54) |
| Valoarea mediană, luni (IÎ 95%) | 17,7 (16,0, 22,4) | 15,3 (13,7, 16,8) |
| RR (IÎ 95%); valoarea pb | 0,73 (0,54, 0,99); p=0,0386 | |
| Rata de răspuns obiectiva | ||
| RRO, % (IÎ 95%) | 64% (55%, 72%) | 36% (30%, 42%) |
| Raportul probabilităților (IÎ 95%); valoarea p | 3,10 (2,00, 4,80); p<0,0001 | |
| Durata răspunsului (DR)a | ||
| Mediana DR (IÎ 95%), luni | 6,90 (5,52, NE) | 5,55 (4,17, 9,56) |
| Pacienți cu DR ≥ 6 luni | 31,9% | 20,0% |
IÎ = Interval de încredere
NE = nu poate fi estimat
Rezultatele SFP, DR și RRO provin din datele din 10 iulie 2023 atunci când s-au efectuat testarea ipotezelor și analiza finală pentru aceste criterii finale de evaluare. Rezultatele privind SG provin din datele din 26 aprilie 2024, din cea de-a doua analiză intermediară privind SG. a
Analiză centrală independentă în regim orb (BICR) b
Valoarea p este comparată cu un nivel bilateral de eficacitate de 0,0142. Astfel rezultatele SG nu sunt semnificative la a doua analiză intermediară.
Figura 3: Curba Kaplan-Meier a SFP la pacienți cu NSCLC tratați anterior, obținută în urma evaluării BICR
Beneficiul SFP al Rybrevant-CP comparativ cu CP a fost constant în toate subgrupurile predefinite analizate, care au inclus etnia, vârsta, sexul, antecedentele de fumător și statusul metastazelor din sistemul nervos central la intrarea în studiu.
Figura 4: Curba Kaplan-Meier a SG la pacienți cu NSCLC tratați anterior
Date privind eficacitatea pentru metastazele intracraniene
Pacienții cu metastaze intracraniene asimptomatice sau tratate anterior și stabile au fost eligibili pentru a fi randomizați în MARIPOSA-2.
Tratamentul cu Rybrevant-CP a fost asociat cu o creștere numerică a RRO intracraniene (23,3% pentru Rybrevant-CP comparativ cu 16,7% pentru CP, risc relativ de 1,52; IÎ 95% (0,51, 4,50) și durata răspunsului intracranian (13,3 luni; IÎ 95% (1,4, NE) în brațul cu Rybrevant-CP, comparativ cu 2,2 luni; IÎ 95% (1,4, NE) în brațul cu CP). Perioada mediană de urmărire pentru Rybrevant-CP a fost de aproximativ 18,6 luni.
Cancer pulmonar fără celule mici (NSCLC) netratat anterior, cu mutații ale inserției Exon 20 (PAPILLON).
PAPILLON este un studiu randomizat, în regim deschis, multicentric, de fază 3, care compară tratamentul cu Rybrevant în asociere cu carboplatină și pemetrexed cu chimioterapia în monoterapie (carboplatină și pemetrexed) la pacienți care nu au fost tratați anterior, cu NSCLC avansat local sau metastazat, cu mutații activatoare ale inserției Exon 20 EGFR. Eșantioanele de țesut tumoral (92,2%) și/sau plasmatic (7,8%) pentru toți cei 308 de pacienți au fost testate la nivel local pentru a determina statusul mutației de inserție Exon 20 EGFR utilizând secvențierea de generație următoare (NGS) la 55,5% dintre pacienți și/sau reacția în lanț a polimerazei (PCR) la 44,5% dintre pacienți. Testarea centrală a fost efectuată, de asemenea, utilizând testul de țesut AmoyDx® LC10, testul țintă Thermo Fisher Oncomine Dx și testul de plasmă Guardant 360® CDx.
Pacienții care au prezentat metastaze cerebrale la screening au fost eligibili pentru participare după ce au fost tratați definitiv, stabili din punct de vedere clinic, asimptomatici și în afara tratamentului cu corticosteroizi timp de cel puțin 2 săptămâni înainte de randomizare.
Rybrevant a fost administrat intravenos în doză de 1400 mg (la pacienți cu greutatea < 80 kg) sau 1750 mg (la pacienți cu greutatea ≥80 kg), o dată pe săptămână, timp de 4 săptămâni, apoi la fiecare 3 săptămâni, în doză de 1750 mg (la pacienți cu greutatea < 80 kg) sau 2100 mg (la pacienți cu greutatea ≥80 kg), începând cu săptămâna 7, până la progresia bolii sau la atingerea unei toxicități inacceptabile. Carboplatina a fost administrată intravenos, la o valoare a ariei de sub curba concentrației plasmatice în funcție de timp de 5 mg/ml pe minut (ASC 5), o dată la 3 săptămâni, timp de până la 12 săptămâni. Pemetrexed a fost administrat intravenos în doză de 500 mg/m2, o dată la 3 săptămâni, până la progresia bolii sau la atingereaunei toxicități inacceptabile. Randomizarea a fost stratificată în funcție de statusul de performanță ECOG (0 sau 1) și de prezența metastazelor cerebrale anterioare (da sau nu). Pacienților randomizați tratați cu carboplatină și pemetrexed, cărora li s-a confirmat progresia bolii, li s-a permis să treacă la administrarea de Rybrevant în monoterapie. În total, au fost randomizați 308 de subiecți (1:1) pentru a li se administra Rybrevant în asociere cu carboplatină și pemetrexed (N=153) sau carboplatină și pemetrexed (N=155). Vârsta mediană a fost de 62 de ani (interval: 27 până la 92 de ani), 39% dintre subiecți având vârsta ≥65 de ani; 58% au fost femei; 61% au fost asiatici și 36% au fost caucazieni. La momentul inițial, scorul de performanță ECOG (Eastern Cooperative Oncology Group) a fost 0 (35%) sau 1 (64%); 58% nu au fumat niciodată; 23% au avut antecedente de metastaze cerebrale și 84% au avut cancer în stadiul IV la diagnosticul inițial.
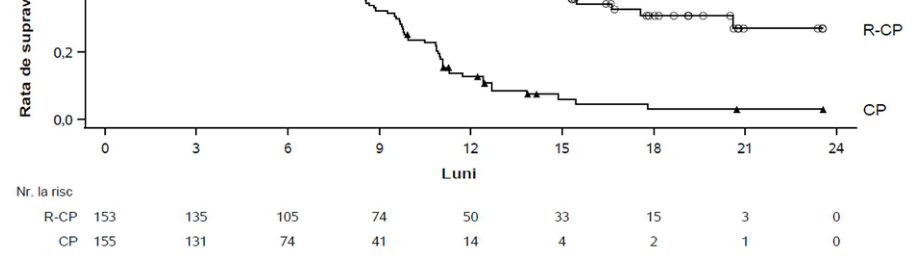
Obiectivul principal pentru PAPILLON a fost supraviețuirea fără progresia bolii (SFP), astfel cum a fost evaluat de BICR. Perioada mediană de monitorizare a fost de 14,9 luni (interval: 0,3 până la 27,0).
Rezultatele eficacității sunt rezumate în tabelul 13.
Tabelul 13: Rezultatele eficacității în PAPILLON
| Rybrevant+ carboplatină+ pemetrexed (N=153) | carboplatină+ pemetrexed (N=155) | |
| Supraviețuire fără progresia bolii (SFP)a | ||
|---|---|---|
| Număr de evenimente | 84 (55%) | 132 (85%) |
| Valoarea mediană, luni (IÎ 95%) | 11,4 (9,8, 13,7) | 6,7 (5,6, 7,3) |
| RR (IÎ 95%); valoarea p | 0,395 (0,29, 0,52); p<0,0001 | |
| Rata de răspuns obiectiva, b | ||
| RRG, % (IÎ 95%) | 73% (65%, 80%) | 47% (39%, 56%) |
| Raportul probabilităților (IÎ 95%); valoarea p | 3,0 (1,8, 4,8); p<0,0001 | |
| Răspuns complet | 3,9% | 0,7% |
| Răspuns parțial | 69% | 47% |
| Supraviețuire globală (SG)c | ||
| Număr de evenimente | 40 | 52 |
| Valoarea mediană SG, luni (IÎ 95%) | NE (28,3, NE) | 28,6 (24,4, NE) |
| RR (IÎ 95%); valoarea p | 0,756 (0,50, 1,14); p=0,1825 | |
IÎ = Interval de încredere
NE = nu poate fi estimat a
Analiză centrală independentă în regim orb (BICR) efectuată de RECIST v1.1 b
Pe baza estimării Kaplan-Meier. c
Pe baza rezultatelor unei SG actualizate cu o perioadă mediană de monitorizare de 20,9 luni. Analiza SG nu a fost ajustată pentru efectele de confuzie potențiale ale schimbării tratamentului (78 [50.3%] de pacienți din grupul cu carboplatină + pemetrexed cărora li s-a administrat ulterior un tratament cu Rybrevant în monoterapie).
Figura 5: Curba Kaplan-Meier a SFP la pacienți cu NSCLC netratați anterior, obținută în urma evaluării BICR

Beneficiul SFP al Rybrevant în asociere cu carboplatină și pemetrexed, comparativ cu carboplatină și pemetrexed, a fost constant în toate subgrupurile predefinite de metastaze cerebrale la intrarea în studiu (da sau nu), vârstă (< 65 sau ≥65), sex (masculin sau feminin), rasă (asiatică sau non-asiatică), greutate (< 80 kg sau ≥80 kg), scor de performanță ECOG (0 sau 1) și antecedente de fumător (da sau nu).
Figura 6: Curba Kaplan-Meier a SG la pacienți cu NSCLC netratați anterior


Cancer pulmonar fără celule mici (NSCLC), tratat anterior, cu mutații ale inserției Exon 20 (CHRYSALIS)
CHRYSALIS este un studiu multicentric, în regim deschis, cu mai multe cohorte, efectuat pentru a evalua siguranța și eficacitatea Rybrevant la pacienții cu NSCLC avansat local sau metastazat. Eficacitatea a fost evaluată la 114 pacienți cu NSCLC avansat local sau metastazat, care prezentau mutații de inserție Exon 20 EGFR, a căror boală progresase în timpul sau după chimioterapia pe bază de platină și care au avut o perioadă mediană de urmărire de 12,5 luni. Eșantioane de țesut tumoral (93%) și/sau plasmatic (10%) pentru toți pacienții au fost testate la nivel local pentru a determina statusul mutației de inserție Exon 20 EGFR utilizând secvențierea de generație următoare (NGS) la 46% dintre pacienți și/sau reacția în lanț a polimerazei (PCR) la 41% dintre pacienți; pentru 4% dintre pacienți, metodele de testare nu au fost specificate. Pacienții care au prezentat în ultimii 2 ani metastaze cerebrale netratate sau cu antecedente de boli pulmonare interstițiale (BPI) care necesită tratament cu steroizi cu acțiune prelungită sau alți agenți imunosupresori nu au fost eligibili pentru studiu. Rybrevant a fost administrat intravenos în doză de 1050 mg la pacienți cu greutatea < 80 kg sau 1400 mg la pacienți cu greutatea ≥80 kg, o dată pe săptămână, timp de 4 săptămâni, apoi la fiecare 2 săptămâni, începând cu săptămâna 5, până la dispariția beneficiului clinic sau a toxicității inacceptabile. Criteriul final principal de evaluare a eficacității a fost rata de răspuns global (RRG) evaluată de investigator, definită ca răspuns complet (RC) sau răspuns parțial (RP) confirmat pe baza RECIST v1.1. În plus, criteriul de evaluare final principal a fost evaluat prin intermediul unei evaluări centrale independente în regim orb (BICR). Criteriile finale secundare de evaluare a eficacității au inclus durata răspunsului (DR).
Vârsta mediană a fost de 62 ani (interval: 36–84) ani, cu 41% dintre pacienți cu vârsta ≥65 ani; 61% au fost femei; și 52% au fost asiatici și 37% au fost caucazieni. Numărul median de tratamente anterioare a fost de 2 (interval: 1 până la 7 terapii). La momentul inițial, 29% din pacienți aveau scorul de performanță ECOG (Eastern Cooperative Oncology Group) de 0 și 70% aveau scorul de performanță ECOG de 1; 57% nu au fumat niciodată; 100% aveau cancer în stadiul IV; iar 25% aveau tratament anterior pentru metastaze cerebrale. Inserțiile din Exon 20 au fost observate la 8 reziduuri diferite; cele mai frecvente reziduuri au fost A767 (22%), S768 (16%), D770 (12%) și N771 (11%).
Rezultatele cu privire la eficacitate sunt prezentate pe scurt în Tabelul 14.
Tabelul 14: Rezultatele eficacității în CHRYSALIS
| Evaluare Investigator (N=114) | |
| Rata de răspuns globala, b(IÎ 95%) | 37% (28%, 46%) |
| Răspuns complet | 0% |
| Răspuns parțial | 37% |
| Durata răspunsului | |
|---|---|
| Valoarea medianăc(IÎ 95%), luni | 12,5 (6,5; 16,1) |
| Pacienți cu DR ≥6 luni | 64% |
IÎ = Interval de încredere a
Răspuns confirmat b
Rezultatele RRG și DR obținute în urma evaluării efectuate de investigator au fost similare cu cele raportate în urma evaluării BICR; valorile RRG obținute de BICR au fost 43% (34%, 53%), cu 3% rata RC și 40% rata RP, DR mediană obținută de BICR a fost 10,8 luni (IÎ 95%: 6,9, 15,0) iar pacienții cu DR ≥ 6 luni conform evaluării BICR au reprezentat 55%. c
Pe baza estimării Kaplan-Meier.
Activitatea antitumorală a fost observată în toate cazurile purtătoare de mutație.
Vârstnici
Nu s-au observat diferențe la modul general în ceea ce privește eficacitatea la pacienții cu vârstă ≥65 ani față de pacienții cu vârstă < 65 ani.
Copii și adolescenți
Agenția Europeană pentru Medicamente a acordat o derogare de la obligația de depunere a rezultatelor studiilor efectuate cu Rybrevant la toate subgrupele de copii și adolescenți în tratamentul cancerului pulmonar fără celule mici (vezi pct. 4.2 pentru informații privind utilizarea la copii și adolescenți).
5.2 Proprietăți farmacocinetice
Pe baza datelor privind tratamentul cu Rybrevant în monoterapie, aria de sub curba concentrației plasmatice în funcție de timp (ASC1 săptămână) a amivantamabului crește proporțional în intervalul de doze de la 350 la 1750 mg.
Pe baza simulărilor din modelul farmacocinetic populațional, ASC1 săptămână a fost de aproximativ 2,8 ori mai mare după cea de-a cincea doză pentru schema de tratament la 2 săptămâni și de 2,6 ori mai mare după cea de-a patra doză pentru schema de tratament la 3 săptămâni. Concentrațiile plasmatice la starea de echilibru ale amivantamab au fost atinse până în săptămâna 13 atât pentru schema de tratament la 3 săptămâni, cât și pentru schema de tratament la 2 săptămâni, iar acumularea sistemică a fost de 1,9 ori.
Distribuție
Pe baza estimărilor parametrilor farmacocinetici individuali ai amivantamab din analiza farmacocinetică populațională, valoarea mediei geometrice (CV%) a volumului total de distribuție este de 5,12 (27,8%) l, după administrarea dozei recomandate de Rybrevant.
Eliminare
Pe baza estimărilor parametrilor farmacocinetici individuali ai amivantamab din analiza farmacocinetică populațională, valoarea mediei geometrice (CV%) a clearance-ului liniar (CL) și a timpului de înjumătățire plasmatică asociat cu clearance-ul liniar este de 0,266 (30,4%) l/zi, respectiv de 13,7 (31,9%) zile.
Categorii speciale de populație
Vârstnici
Nu s-au observat diferențe semnificative din punct de vedere clinic în ceea ce privește farmacocinetica amivantamab în funcție de vârstă (21-88 de ani).
Insuficiență renală
Nu s-a observat niciun efect semnificativ din punct de vedere clinic asupra farmacocineticii amivantamab la pacienții cu insuficiență renală ușoară (60 ≤ clearance-ul creatininei [ClCr] < 90 ml/min), moderată (29 ≤ ClCr < 60 ml/min) sau severă (15 ≤ CrCl < 29 ml/min). Datele referitoare la pacienții cu insuficiență renală severă sunt limitate (n=1), dar nu există dovezi care să sugereze necesitatea ajustării dozei la acești pacienți. Nu se cunoaște efectul bolii renale în stadiu terminal (CrCl < 15 ml/min) asupra farmacocineticii amivantamabului.
Insuficiență hepatică
Este puțin probabil ca modificările funcției hepatice să aibă vreun efect asupra eliminării amivantamabului, deoarece moleculele pe bază de IgG1, cum este amivantamabul, nu sunt metabolizate pe cale hepatică.
Nu s-a observat niciun efect semnificativ din punct de vedere clinic asupra farmacocineticii amivantamab pe baza insuficienței hepatice ușoare [(bilirubina totală ≤LSVN și AST > LSVN) sau (LSVN < bilirubina totală ≤1,5 x LSVN)] sau moderate (1,5×LSVN < bilirubina totală ≤3×LSVN și orice AST). Datele referitoare la pacienții cu insuficiență hepatică moderată sunt limitate (n=1), dar nu există dovezi care să sugereze necesitatea ajustării dozei la acești pacienți. Nu se cunoaște efectul insuficienței hepatice severe (bilirubină totală > 3 ori LSVN) asupra farmacocineticii amivantamabului.
Copii și adolescenți
Nu s-a studiat farmacocinetica Rybrevant la copii și adolescenți.
5.3 Date preclinice de siguranță
Datele non-clinice nu au evidențiat niciun risc special pentru om pe baza studiilor convenționale farmacologice privind toxicitatea după doze repetate.
Carcinogenitate și mutagenitate
Nu au fost efectuate studii la animale pentru a stabili potențialul carcinogen al amivantamabului. Studiile de genotoxicitate și carcinogenitate de rutină nu sunt, în general, aplicabile produselor farmaceutice biologice, deoarece proteinele mari nu pot difuza în celule și nu pot interacționa cu ADN-ul sau materialul cromozomial.
Toxicitate asupra funcției de reproducere
Nu au fost efectuate studii la animale pentru a evalua efectele asupra reproducerii și dezvoltării fetale; cu toate acestea, pe baza mecanismului său de acțiune, amivantamabul poate cauza leziuni fetale sau anomalii de dezvoltare. Așa cum este raportat în literatura de specialitate, reducerea, eliminarea sau întreruperea semnalizării EGFR embrionare sau materne pot preveni implantarea, pot cauza pierderea embrionului fetal în timpul diferitelor etape ale gestației (prin efecte asupra dezvoltării placentare), pot cauza anomalii de dezvoltare la nivelul mai multor organe sau moartea precoce la fetușii supraviețuitori. În mod similar, inactivarea TME sau a factorului său de creștere hepatocit ligand (HGF) a fost embrionar letal datorită defectelor severe în dezvoltarea placentară, iar fetușii au prezentat defecte în dezvoltarea musculară la nivelul mai multor organe. Se cunoaște că IgG1 uman traversează placenta; prin urmare, amivantamabul are potențial de a fi transmis de la mamă la fătul în dezvoltare.
6. PROPRIETĂȚI FARMACEUTICE
6.1 Lista excipienților
Sare disodică a acidului etilendiaminotetraacetic (EDTA) dihidrat
L-histidină
Clorhidrat de L-histidină monohidrat
L-metionină
Polisorbat 80 (E433)
Zahăr
Apă pentru preparate injectabile
6.2 Incompatibilități
Acest medicament nu trebuie amestecat cu alte medicamente, cu excepția celor menționate la pct. 6.6.
6.3 Perioada de valabilitate
Flacon nedeschis
3 ani
După diluare
Stabilitatea chimică și fizică în uz a fost demonstrată pentru o perioadă de 10 ore la temperaturi de 15°C până la 25°C la lumina camerei. Din punct de vedere microbiologic, cu excepția situației în care metoda de diluție a soluției exclude riscul de contaminare microbiană, produsul trebuie administrat imediat. Dacă soluțiile nu sunt utilizate imediat, responsabilitatea în ceea ce privește timpul și condițiile de folosire revine utilizatorului.
6.4 Precauții speciale pentru păstrare
A se păstra la frigider (2°C la 8°C).
A nu se congela.
A se păstra în ambalajul original, pentru a fi protejat de lumină.
Pentru condițiile de păstrare după diluția medicamentului, vezi pct. 6.3.
6.5 Natura și conținutul ambalajului
7 ml de concentrat într-un flacon din sticlă de tip 1 cu capac din elastomer și capsă din aluminiu cu cap detașabil, conținând amivantamab 350 mg. Pachet cu 1 flacon.
6.6 Precauții speciale pentru eliminarea reziduurilor și alte instrucțiuni de manipulare
A se prepara soluția pentru perfuzie intravenoasă utilizând o tehnică aseptică după cum urmează:
Preparare
- A se determina doza necesară și numărul de flacoane de Rybrevant necesare în funcție de greutatea inițială a pacientului (vezi pct. 4.2). Fiecare flacon conține amivantamab 350 mg.
- Pentru tratamentul la fiecare 2 săptămâni, pacienților cu greutate corporală < 80 kg li se administrează 1050 mg, iar pacienților cu greutate corporală ≥80 kg li se administrează 1400 mg, o dată pe săptămână, în total 4 doze, apoi la fiecare 2 săptămâni, începând cu săptămâna 5.
- Pentru tratamentul la fiecare 3 săptămâni, pacienților cu greutate corporală < 80 kg li se administrează 1400 mg, o dată pe săptămână, în total 4 doze, apoi 1750 mg la fiecare 3 săptămâni începând cu săptămâna 7, iar pacienților cu greutate corporală ≥80 kg li se administrează 1750 mg, o dată pe săptămână, în total 4 doze, apoi 2100 mg la fiecare 3 săptămâni, începând cu săptămâna 7.
- A se verifica dacă soluția de Rybrevant este incoloră până la galben deschis. A nu se utiliza în caz de decolorare sau dacă sunt prezente particule vizibile.
- A se extrage și apoi a se arunca un volum de soluție injectabilă de glucoză 5% sau soluție injectabilă de clorură de sodiu 9 mg/ml (0,9%) din punga de perfuzie de 250 ml, care este egal cu volumul necesar de soluție de Rybrevant care trebuie adăugată (aruncați 7 ml de solvent din punga de perfuzie pentru fiecare flacon). Pungile de perfuzie trebuie să fie fabricate din clorură de polivinil (PVC), polipropilenă (PP), polietilenă (PE) sau amestec poliolefinic (PP+PE).
- A se extrage 7 ml de Rybrevant din fiecare flacon necesar, apoi adăugați-l în punga de perfuzie. Fiecare flacon este umplut în exces cu 0,5 ml pentru a asigura un volum extractibil suficient. Volumul final din punga de perfuzie trebuie să fie de 250 ml. A se arunca orice cantitate neutilizată rămasă în flacon.
- A se răsturna ușor punga de perfuzie pentru a amesteca soluția. A nu se agita.
- A se inspecta vizual înainte de administrare, pentru detectarea conținutului de particule și a modificărilor de culoare. A nu se utiliza în caz de decolorare sau dacă se observă particule vizibile.
Administrare
- A se administra soluția diluată prin perfuzie intravenoasă utilizând un set de perfuzie prevăzut cu un regulator de debit și cu un filtru de polietersulfonă (PES) în linie, steril, apirogen, cu capacitate redusă de fixare a proteinelor (dimensiunea porilor de 0,22 sau 0,2 micrometri). Seturile de administrare trebuie realizate fie din poliuretan (PU), polibutadienă (PBD), PVC, PP sau PE.
- Înainte de inițierea fiecărei perfuzii cu Rybrevant, setul de administrare cu filtru trebuie pregătit fie cu soluție de glucoză 5%, fie cu soluție de clorură de sodiu 0,9%.
- A nu se perfuza Rybrevant concomitent cu alte medicamente prin intermediul aceleiași linii intravenoase.
- Soluția diluată trebuie administrată în decurs de 10 ore (inclusiv timpul de perfuzare) la temperatura camerei (15 °C până la 25 °C) și la lumina camerei.
- Datorită apariției frecvente a reacțiilor adverse legate de perfuzie la momentul administrării primei doze, amivantamab trebuie administrat prin intermediul unei căi venoase periferice în Săptămâna 1 și Săptămâna 2; administrarea perfuzabilă pe cale centrală se va efectua în următoarele săptămâni când riscul de RALP este mai mic. Vezi vitezele de perfuzare la pct. 4.2.
Eliminare
Acest medicament este de unică folosință și orice medicament neutilizat care nu este administrat în decurs de 10 ore trebuie eliminat în conformitate cu reglementările locale.
7. DEȚINĂTORUL AUTORIZAȚIEI DE PUNERE PE PIAȚĂ
Janssen-Cilag International NV
Turnhoutseweg 30
B-2340 Beerse
Belgia
8. NUMĂRUL(ELE) AUTORIZAȚIEI DE PUNERE PE PIAȚĂ
EU/1/21/1594/001
9. DATA PRIMEI AUTORIZĂRI SAU A REÎNNOIRII AUTORIZAȚIEI
Data primei autorizări: 9 decembrie 2021
Data ultimei reînnoiri a autorizației: 11 septembrie 2023
10. DATA REVIZUIRII TEXTULUI
Informații detaliate privind acest medicament sunt disponibile pe site-ul Agenției Europene pentru Medicamente https://www.ema.europa.eu.
Acest medicament face obiectul unei monitorizări suplimentare. Acest lucru va permite identificarea rapidă de noi informații referitoare la siguranță. Profesioniștii din domeniul sănătății sunt rugați să raporteze orice reacții adverse suspectate. Vezi pct. 4.8 pentru modul de raportare a reacțiilor adverse.
1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI
Rybrevant 1600 mg soluție injectabilă
Rybrevant 2240 mg soluție injectabilă
2. COMPOZIȚIA CALITATIVǍ ȘI CANTITATIVǍ
Rybrevant 1600 mg soluție injectabilă
Un ml de soluție injectabilă conține 160 mg de amivantamab.
Un flacon de 10 ml soluție injectabilă conține 1600 mg de amivantamab.
Rybrevant 2240 mg soluție injectabilă
Un ml de soluție injectabilă conține 160 mg de amivantamab.
Un flacon de 14 ml soluție injectabilă conține 2240 mg de amivantamab.
Amivantamab este un anticorp bispecific complet uman, pe bază de imunoglobulină G1 (IgG1), care vizează factorul de creștere epidermică (EGF) și receptorii de tranziție mezenchimal-epidermică (TME), produși de o linie de celule ale mamiferelor (ovar de hamster chinezesc [CHO]) utilizând tehnologia ADN-ului recombinant.
Excipienți cu efect cunoscut:
Un ml de soluție conține 0,6 mg de polisorbat 80.
Pentru lista tuturor excipienților, vezi pct. 6.1.
3. FORMA FARMACEUTICĂ
Soluție injectabilă.
Soluția este de la incoloră până la galben deschis.
4. DATE CLINICE
4.1 Indicații terapeutice
Rybrevant formă farmaceutică cu administrare subcutanată este indicat:
- în asociere cu lazertinib pentru tratamentul de primă linie la pacienții adulți cu cancer pulmonar fără celule mici (NSCLC) în stadiu avansat, cu deleții în Exonul 19 al EGFR sau cu mutații de substituție în Exonul 21 L858R.
- în monoterapie, pentru tratamentul pacienților adulți cu cancer pulmonar fără celule mici (NSCLC) în stadiu avansat, cu mutații activatoare ale inserției Exon 20 a EGFR, după eșecul terapiei pe bază de platină.
4.2 Doze și mod de administrare
Tratamentul cu Rybrevant formă farmaceutică cu administrare subcutanată trebuie inițiat și supervizat de un medic cu experiență în utilizarea medicamentelor antineoplazice.
Înaintea inițierii tratamentului cu Rybrevant formă farmaceutică cu administrare subcutanată, trebuie determinată existența mutației EGFR în eșantioanele de țesut tumoral sau plasmatic utilizând o metodă de testare validată. Dacă nu se detectează nicio mutație în eșantionul plasmatic, trebuie testat dacă țesutul tumoral este disponibil într-o cantitate suficientă și are o calitate adecvată, din cauza potențialului de rezultate fals-negative ale unui test bazat pe plasmă. Nu este necesară repetarea testării odată ce statusul mutației EGFR a fost stabilit (vezi pct. 5.1).
Rybrevant formă farmaceutică cu administrare subcutanată trebuie administrat de către un profesionist din domeniul sănătății, care să aibă acces la asistență medicală adecvată pentru abordarea terapeutică a reacțiilor adverse legate de administrare, dacă apar.
Doze
Trebuie administrate medicații prealabile pentru a reduce riscul de reacții adverse legate de administrare asociate cu Rybrevant formă farmaceutică cu administrare subcutanată (vezi mai jos „Ajustările dozei” și „Medicația concomitentă recomandată”).
Dozele recomandate de Rybrevant, atunci când este utilizat în asociere cu lazertinib sau în monoterapie, în funcție de greutatea corporală a pacientului la momentul inițial, sunt prezentate în Tabelul 1.
Tabelul 1: Doza recomandată de Rybrevant formă farmaceutică cu administrare subcutanată
| Greutatea corporală a pacientului la momentul inițial* | Doza recomandată | Schemă |
|---|---|---|
| Mai mică de 80 kg | 1600 mg |
|
| Mai mare sau egală cu 80 kg | 2240 mg |
|
* Ajustările dozei nu sunt necesare pentru modificările ulterioare ale greutății corporale.
Dacă se administrează în asociere cu lazertinib, se recomandă administrarea Rybrevant formă farmaceutică cu administrare subcutanată în orice moment după administrarea lazertinib, dacă sunt adminstrate în aceeași zi. Consultați pct. 4.2 din Rezumatul caracteristicilor produsului pentru lazertinib pentru informațiile privind dozele recomandate de lazertinib.
Durata tratamentului
Se recomandă ca pacienților să li de administreze Rybrevant formă farmaceutică cu administrare subcutanată până la progresia bolii sau până la apariția toxicității inacceptabile.
Doza omisă
Dacă se omite o doză de Rybrevant formă farmaceutică cu administrare subcutanată între săptămâna 1 și săptămâna 4, aceasta trebuie administrată în decurs de 24 de ore. Dacă se omite o doză de Rybrevant formă farmaceutică cu administrare subcutanată începând cu săptămâna 5, aceasta trebuie administrată în decurs de 7 zile. În caz contrar, doza omisă nu trebuie administrată, iar următoarea doză trebuie administrată conform schemei de administrare obișnuite.
Ajustările dozei
În cazul reacțiilor adverse de grad 3 sau 4, administrarea trebuie întreruptă până la momentul ameliorării reacțiilor adverse până la reacții adverse de grad ≤1 sau revenirea la starea inițială. Dacă o întrerupere durează 7 zile sau mai puțin, reîncepeți cu doza curentă. Dacă o întrerupere durează mai mult de 7 zile, se recomandă reînceperea tratamentului cu o doză redusă, așa cum este prezentat în Tabelul 2. După Tabelul 2 sunt prezentate, de asemenea, și modificările specifice ale dozei în funcție de reacțiile adverse specifice.
Dacă se administrează în asociere cu lazertinib, consultați pct. 4.2 din Rezumatul caracteristicilor produsului pentru lazertinib pentru informații privind ajustările dozei.
Tabelul 2: Recomandări privind modificarea dozei în cazul apariției reacțiilor adverse
| Doza* | Doza după prima întrerupere determinată de apariția reacțiilor adverse | Doza după a doua întrerupere determinată de apariția reacțiilor adverse | Doză după a treia întrerupere determinată de apariția reacțiilor adverse |
|---|---|---|---|
| 1600 mg | 1050 mg | 700 mg | Se oprește tratamentul cu Rybrevant formă farmaceutică cu administrare subcutanată |
| 2240 mg | 1600 mg | 1050 mg |
* Doza la care a survenit reacția adversă.
Reacții adverse legate de administrare
Trebuie administrate medicații prealabile pentru a reduce riscul de reacții adverse legate de administrare asociate cu Rybrevant formă farmaceutică cu administrare subcutanată (vezi „Medicația concomitentă recomandată”). Injecțiile trebuie întrerupte la primul semn de reacții adverse legate de administrare. Medicamente suplimentare de susținere (de exemplu, glucocorticoizi, antihistaminice, antipiretice și antiemetice suplimentare) trebuie administrate conform indicațiilor clinice (vezi pct. 4.4).
- Grad 1-3 ( ușor-sever): Odată cu recuperarea în urma simptomelor, se reia administrarea injectabilă a Rybrevant formă farmaceutică cu administrare subcutanată. Medicamentele administrate concomitent trebuie administrate cu următoarea doză, inclusiv dexametazonă (20 mg) sau echivalent (vezi Tabelul 3).
- Recidivă de grad 3 sau grad 4 (cu potențial letal): Întrerupeți definitiv tratamentul cu Rybrevant.
Evenimente tromboembolice venoase (TEV) asociate cu administrarea în asociere cu lazertinib La inițierea tratamentului, trebuie administrate profilactic anticoagulante, pentru a preveni evenimentele TEV la pacienții cărora li se administrează Rybrevant formă farmaceutică cu administrare subcutanată în asociere cu lazertinib. Conform ghidurilor clinice, pacienților trebuie să li se administreze tratament profilactic fie cu un anticoagulant oral cu acțiune directă (AOAD), fie cu o heparină cu masă moleculară mică (HMMM). Utilizarea antagoniștilor vitaminei K nu este recomandată.
Pentru evenimentele TEV asociate cu instabilitate clinică (de ex. insuficiență respiratorie sau disfuncție cardiacă), administrarea ambelor medicamente trebuie oprită până când pacientul este stabil clinic. După aceea, administrarea ambelor medicamente poate fi reluată în aceleași doze. În cazul recurenței, în ciuda tratamentului adecvat cu anticoagulant, se întrerupe administrarea Rybrevant. Tratamentul poate continua cu lazertinib în aceeași doză (vezi pct. 4.4).
Reacții cutanate și unghiale
Se recomandă terapia profilactică cu antibiotice cu administrare orală și topică pentru a reduce riscul de apariție și severitatea reacțiilor la nivelul pielii si unghiilor la pacienții cărora li se administrează Rybrevant. Se recomandă, de asemenea, utilizarea unei creme hidratante necomedogene (de preferință pe bază de ceramide sau alte formule care asigură hidratarea îndelungată a pielii și nu conțin agenți de uscare) pe față și pe întreg corpul (cu excepția scalpului) și a unei soluții de clorhexidină pentru spălarea mâinilor și a picioarelor. Pacienții trebuie sfătuiți să limiteze expunerea la soare pe durata tratamentului cu Rybrevant și timp de 2 luni după acesta. Pentru informații suplimentare despre profilaxia reacțiilor cutanate și unghiale, vezi pct. 4.4.
Dacă pacientul dezvoltă o reacție cutanată sau unghială de grad 1-2, trebuie inițiată terapia de susținere, conform indicațiilor clinice; dacă nu există nicio ameliorare după 2 săptămâni, pentru erupția cutanată persistentă de gradul 2, trebuie luată în considerare reducerea dozei (vezi Tabelul 2). Dacă pacientul dezvoltă o reacție cutanată sau unghială de grad 3, trebuie inițiat tratamentul de susținere, conform indicațiilor clinice și trebuie luată în considerare întreruperea tratamentului cu Rybrevant formă farmaceutică cu administrare subcutanată până la ameliorarea reacției adverse. După dispariția reacției cutanate sau unghiale ≤grad 2, tratamentul cu Rybrevant formă farmaceutică cu administrare subcutanată trebuie reluat cu o doză redusă. Dacă pacientul dezvoltă reacții cutanate de gradul 4, tratamentul cu Rybrevant trebuie întrerupt permanent (vezi pct. 4.4).
Boala pulmonară interstițială
Tratamentul cu Rybrevant formă farmaceutică cu administrare subcutanată trebuie oprit dacă se suspectează boală pulmonară interstițială (BPI) sau reacții adverse asemănătoare BPI (pneumonită). Dacă se confirmă că pacientul a dezvoltat BPI sau reacții adverse similare BPI (de exemplu, pneumonită), tratamentul cu Rybrevant trebuie oprit permanent (vezi pct. 4.4).
Medicația concomitentă recomandată
Înainte de doza inițială (săptămâna 1, ziua 1), trebuie administrate antihistaminice, antipiretice și glucocorticoizi pentru a reduce riscul de reacții adverse legate de administrare (vezi Tabelul 3). Pentru dozele ulterioare, este necesară administrarea de antihistaminice și antipiretice. După o perioadă prelungită de întrerupere a administrării dozei, trebuie reluată de asemenea administrarea de glucocorticoizi. Trebuie administrate antiemetice, dacă este necesar.
Tabelul 3: Schema de administrare a premedicației
| Medicație prealabilă | Doză | Cale de administrare | Interval recomandat de administrare înainte de administrarea de Rybrevant formă farmaceutică cu administrare subcutanată |
|---|---|---|---|
| Antihistaminic* | Difenhidramină (25 până la 50 mg) sau echivalent | Intravenoasă | 15 până la 30 de minute |
| Orală | 30 până la 60 de minute | ||
| Antipiretic* | Paracetamol/Acetaminofen (650 până la 1000 mg) sau echivalent | Intravenoasă | 15 până la 30 de minute |
| Orală | 30 până la 60 de minute | ||
| Glucocorticoid† | Dexametazonă (20 mg) sau echivalent | Intravenoasă | 45 până la 60 de minute |
| Orală | Cel puțin 60 de minute | ||
| Glucocorticoid‡ | Dexametazonă (10 mg) sau echivalent | Intravenoasă | 45 până la 60 de minute |
| Orală | 60 până la 90 de minute |
* Necesar la toate dozele.
† Necesar la doza inițială (săptămâna 1, ziua 1) sau la următoarea doză ulterioară în cazul unei reacții adverse legate de administrare.
‡ Opțional pentru dozele ulterioare.
Categorii speciale de populație
Copii și adolescenți
Utilizarea amivantamab nu se justifică la copii și adolescenți în tratamentul cancerului pulmonar fără celule mici.
Vârstnici
Nu sunt necesare ajustări ale dozei (vezi pct. 4.8, pct. 5.1 și pct. 5.2).
Insuficiență renală
Nu s-au efectuat studii specifice cu amivantamab la pacienți cu insuficiență renală. Pe baza analizelor de farmacocinetică (FC) populațională, nu este necesară ajustarea dozei la pacienții cu insuficiență renală ușoară sau moderată. Este necesară prudență la pacienții cu insuficiență renală severă, deoarece amivantamabul nu a fost studiat la această grupă de pacienți (vezi pct. 5.2). În cazul inițierii tratamentului, pacienții trebuie monitorizați pentru depistarea reacțiilor adverse, cu modificări ale dozei, conform recomandărilor de mai sus.
Insuficiență hepatică
Nu s-au efectuat studii specifice cu amivantamab la pacienți cu insuficiență hepatică. Pe baza analizelor de farmacocinetică populațională, nu este necesară ajustarea dozei la pacienții cu insuficiență hepatică ușoară. Este necesară prudență la pacienții cu insuficiență hepatică moderată sau severă, deoarece amivantamab nu a fost studiat la această grupă de pacienți (vezi pct. 5.2). În cazul inițierii tratamentului, pacienții trebuie monitorizați pentru apariția reacțiilor adverse, cu ajustări ale dozei, conform recomandărilor de mai sus.
Mod de administrare
Rybrevant soluție injectabilă este doar pentru administrare sucutanată.
Rybrevant formă farmaceutică cu administrare subcutanată nu este destinat administrării intravenoase și trebuie administrat doar prin injecție subcutanată, în dozele specificate. A se vedea pct. 6.6 pentru instrucțiuni privind manipularea medicamentului înainte de administrare.
Se injectează volumul recomandat de Rybrevant formă farmaceutică cu administrare subcutanată în țesutul subcutanat de la nivelul abdomenului, într-un interval de aproximativ 5 minute. Nu se administrează în alte zone ale corpului deoarece nu există date disponibile.
Dacă pacientul resimte durere, se face o pauză sau se micșorează viteza de administrare. În cazul în care durerea nu se ameliorează prin întreruperea administrării sau prin micșorarea vitezei de administrare, se poate alege un al doilea loc de injectare, pe partea opusă a abdomenului, pentru a administra restul dozei.
În cazul administrării prin intermediul unui set de perfuzie subcutanată, asigurați-vă că întreaga doză de medicament este administrată prin setul de perfuzie. Se poate utiliza soluție de clorură de sodiu 9 mg/ml (0,9%) pentru a împinge produsul medicamentos rămas de-a lungul liniei de perfuzie.
Injecția nu se administrează la nivelul tatuajelor sau cicatricilor sau în zonele în care pielea este de culoare roșie, cu echimoze, sensibilă, dură, nu este intactă sau la mai puțin de 5 cm periombilical. Locurile de injectare trebuie alternate în cazul injecțiilor succesive.
4.3 Contraindicații
Hipersensibilitate la substanța(ele) activă(e) sau la oricare dintre excipienții enumerați la pct. 6.1.
4.4 Atenționări și precauții speciale pentru utilizare
Trasabilitate
Pentru a avea sub control trasabilitatea medicamentelor biologice, numele și numărul lotului medicamentului administrat trebuie înregistrate cu atenție.
Reacții adverse legate de administrare
Reacții adverse legate de administrare au apărut la pacienții tratați cu Rybrevant formă farmaceutică cu administrare subcutanată (vezi pct. 4.8).
Înainte de injecția inițială (săptămâna 1, ziua 1), trebuie administrate antihistaminice, antipiretice și glucocorticoizi pentru a reduce riscul de reacții adverse legate de administrare. Pentru dozele ulterioare, trebuie administrate antihistaminice și antipiretice.
Pacienții trebuie tratați în unități medicale adecvate suportului terapeutic al reacțiilor adverse legate de administrare. Injecțiile trebuie întrerupte la primul semn de reacții adverse legate de administrare de orice severitate, dacă sunt în curs de administrare, iar medicamentele administrate după injecție trebuie administrate conform indicațiilor clinice. După remisiunea simptomelor, injecția trebuie reluată. În cazul reacțiilor adverse legate de administrare de grad 4 sau recurente de grad 3, tratamentul cu Rybrevant formă farmaceutică cu administrare subcutanată trebuie întrerupt permanent (vezi pct. 4.2).
Boala pulmonară interstițială
La pacienții cărora li s-a administrat amivantamab au fost raportate boli pulmonare interstițiale (BPI) sau reacții adverse similare BPI (de exemplu, pneumonită), inclusiv evenimente cu evoluție letală (vezi pct. 4.8). Pacienții trebuie monitorizați pentru simptome care indică BPI/pneumonită (de exemplu, dispnee, tuse, febră). Dacă apar simptome, tratamentul cu Rybrevant trebuie întrerupt până la evaluarea acestor simptome. Trebuie evaluată BPI suspectată sau reacțiile adverse asemănătoare BPI și, dacă este cazul, trebuie inițiat tratamentul adecvat. Rybrevant trebuie întrerupt definitiv la pacienții cu BPI confirmată sau reacții adverse asemănătoare BPI (vezi pct. 4.2).
Evenimente tromboembolice venoase (TEV) în contextul administrării în asociere cu lazertinib
La pacienții cărora li s-a administrat amivantamab în asociere cu lazertinib au fost raportate evenimente tromboembolice venoase (TEV), inclusiv tromboză venoasă profundă (TVP) și embolie pulmonară (EP) (vezi pct. 4.8). Evenimente cu evoluție letală au fost observate în cazul amivantamab formă farmaceutică cu administrare intravenoasă. Conform ghidurilor clinice, pacienților trebuie să li se administreze preventiv fie un anticoagulant oral cu acțiune directă (AOAD), fie o heparină cu masă moleculară mică (HMMM). Utilizarea antagoniștilor vitaminei K nu este recomandată.
Semnele și simptomele de evenimente TEV trebuie monitorizate. Pacienții cu evenimente TEV trebuie tratați cu anticoagulante după cum este indicat clinic. Pentru evenimentele TEV asociate cu instabilitate clinică, tratamentul trebuie oprit până când pacientul este stabil clinic. După aceea, ambele medicamente pot fi reluate în aceeași doză.
În cazul recurenței apărute în ciuda tratamentului adecvat cu anticoagulant, tratamentul cu Rybrevant trebuie oprit definitiv. Tratamentul poate continua cu lazertinib în aceeași doză (vezi pct. 4.2).
Reacții cutanate și unghiale
La pacienții cărora li se administrează amivantamab au apărut erupții cutanate (inclusiv dermatită acneiformă), prurit, xerodermie și ulcerații cutanate (vezi pct. 4.8). Pacienții trebuie instruiți să limiteze expunerea la soare în timpul și timp de 2 luni după tratamentul cu Rybrevant. Se recomandă echipament de protecție și utilizarea de creme cu factor protecție solară cu spectru larg UVA/UVB. Este recomandată o abordare profilactică pentru prevenirea erupțiilor cutanate. Aceasta include terapie profilactică, la inițierea tratamentului, cu un antibiotic cu utilizare orală (de ex. doxiciclină sau minociclină, 100 mg, de două ori pe zi) începând cu Ziua 1, în primele 12 săptămâni de tratament, iar după finalizarea terapiei cu antibiotic cu utilizare orală, aplicarea topică a unei loțiuni cu antibiotic pe scalp (de ex. clindamicină 1%) pentru următoarele 9 luni de tratament. Este recomandată utilizarea unei creme hidratante necomedogene (sunt preferate formulele pe bază de ceramide sau alte formule care asigură hidratarea îndelungată a pielii și nu conțin agenți de uscare) pe față și pe întregul corp (cu excepția scalpului) și a unei soluții de clorhexidină pentru spălarea mâinilor și a picioarelor începând în Ziua 1 și continuând pe toată durata tratamentului.
Se recomandă ca, la momentul administrării dozei inițiale, să fie disponibile prescripții pentru antibiotice de uz topic și/sau cu administrare orală și corticosteroizi de uz topic pentru a reduce la minimum orice întârziere în abordarea terapeutică a erupției cutanate tranzitorii, în cazul în care aceasta se manifestă în ciuda tratamentului profilactic. Dacă apar reacții cutanate, trebuie administrată terapie de susținere, precum și corticosteroizi topici și antibiotice cu utilizare topică și/sau orală. În cazul evenimentelor de grad 3 sau al celor slab tolerate de grad 2, trebuie administrate, de asemenea, antibiotice sistemice și corticosteroizi cu administrare orală. Pacienții care prezintă erupții cutanate severe care au un aspect sau o distribuție atipică sau care nu prezintă o ameliorare în decurs de 2 săptămâni trebuie să se adreseze imediat unui dermatolog. Rybrevant trebuie redus, întrerupt sau întrerupt permanent în funcție de severitate (vezi pct. 4.2).
S-a raportat apariția necrolizei epidermice toxice (NET). Tratamentul cu acest medicament trebuie întrerupt dacă se confirmă NET.
Tulburări oculare
La pacienții cărora li se administrează amivantamab au apărut tulburări oculare, inclusiv cheratită (vezi pct. 4.8). Pacienții care prezintă agravarea simptomelor oculare trebuie îndrumați imediat către un oftalmolog și trebuie să întrerupă utilizarea lentilelor de contact până la evaluarea simptomelor. Vezi pct. 4.2 pentru modificările de doză în cazul reacțiilor adverse oculare de grad 3 sau 4.
Conținutul de sodiu
Acest medicament conține mai puțin de 1 mmol (23 mg) de sodiu per doză, adică practic „nu conține sodiu“ (vezi pct. 6.6).
Conținutul de polisorbat
Acest medicament conține 0,6 mg de polisorbat 80 în fiecare ml, echivalent cu 6 mg per flacon de 10 ml sau cu 8,4 mg per flacon de 14 ml. Polisorbații pot cauza reacții de hipersensibilitate.
4.5 Interacțiuni cu alte medicamente și alte forme de interacțiune
Nu s-au efectuat studii privind interacțiunile cu alte medicamente. Ca anticorp monoclonal IgG1, este puțin probabil ca excreția renală și metabolizarea mediată prin intermediul enzimelor hepatice a amivantamabului nemodificat să fie căi majore de eliminare. Ca atare, nu se așteaptă ca variațiile enzimelor de metabolizare a medicamentului să afecteze eliminarea amivantamab. Datorită afinității mari față de un epitop unic al EGFR și TME, nu se anticipează ca amivantamab să modifice enzimele care metabolizează medicamentul.
Vaccinare
Nu există informații clinice disponibile legate de eficacitatea și profilul de siguranță ale administrării vaccinurilor la pacienții cărora li se administrează amivantamab. Evitați administrarea de vaccinuri cu virusuri vii sau cu virusuri vii-atenuate la pacienții cărora li se administrează amivantamab.
4.6 Fertilitatea, sarcina și alăptarea
Femeile aflate la vârsta fertilă/Contracepția
Femeile aflate la vârsta fertilă trebuie să utilizeze măsuri contraceptive eficace în timpul și până la 3 luni după întreruperea tratamentului cu amivantamab.
Sarcina
Nu există date la om pentru a evalua riscul utilizării amivantamab în timpul sarcinii. Nu au fost efectuate studii de reproducere pe animale pentru a identifica un risc asociat medicamentului. Administrarea moleculelor inhibitoare de TME și EGFR la animalele gestante a determinat o incidență crescută a afectării dezvoltării embrio-fetale, a mortalității embrionare și a avortului. Prin urmare, pe baza mecanismului său de acțiune și a rezultatelor obținute pe modele animale, amivantamab poate dăuna fătului atunci când este administrat unei femei gravide. Amivantamabul nu trebuie administrat în timpul sarcinii, cu excepția cazului în care se consideră că beneficiul tratamentului cu amivantamab depășește riscurile potențiale pentru făt. Dacă pacienta rămâne gravidă în timpul tratamentului cu acest medicament, pacienta trebuie informată cu privire la riscul potențial pentru făt (vezi pct. 5.3).
Alăptarea
Nu se cunoaște dacă amivantamabul este excretat în laptele matern uman. Se știe că IgG uman este excretat în laptele matern în primele câteva zile după naștere, concentrația acestuia scăzând ulterior până la valori joase. În timpul acestei scurte perioade nu poate fi exclus un anumit risc pentru copilul alăptat la sân, deși imunoglubulinele de tip IgG sunt degradate în tractul gastro-intestinal al sugarului alăptat la sân și, astfel, nu sunt absorbite. Trebuie luată o decizie fie de a întrerupe alăptarea, fie de a întrerupe/a omite tratamentul cu amivantamab, având în vedere beneficiul alăptării pentru copil și beneficiul tratamentului pentru femeie.
Fertilitatea
Nu sunt disponibile date pentru determinarea efectelor amivantamabului asupra fertilității la om. Efectele asupra fertilității la bărbați și femei nu au fost evaluate în cadrul unor studii efectuate pe animale.
4.7 Efecte asupra capacității de a conduce vehicule și de a folosi utilaje
Rybrevant poate avea o influență moderată asupra capacității de a conduce vehicule și de a folosi utilaje. Vă rugăm să consultați pct. 4.8 (de ex., amețeală, fatigabilitate, tulburări de vedere). Dacă în urma tratamentului pacienții prezintă simptome, inclusiv reacții adverse legate de vedere, care le afectează capacitatea de concentrare și reacție, acestora li se recomandă să nu conducă sau să nu folosească utilaje până la dispariția efectului medicamentului.
4.8 Reacții adverse
Rezumatul profilului de siguranță
Rybrevant în monoterapie
În setul de date referitoare la Rybrevant formă farmaceutică cu administrare intravenoasă, administrat în monoterapie (N=380), cele mai frecvente reacții adverse de toate gradele au fost erupții cutanate tranzitorii (76%), reacții adverse legate de perfuzie (67%), toxicitate unghială (47%), hipoalbuminemie (31%), edem (26%), fatigabilitate (26%), stomatită (24%), greață (23%) și constipație (23%). Reacțiile adverse grave au inclus BPI (1,3%), RALP (1,1%) și erupții cutanate (1,1%). Trei procente dintre pacienți au întrerupt tratamentul cu Rybrevant din cauza reacțiilor adverse. Cele mai frecvente reacții adverse care au dus la întreruperea tratamentului au fost RALP (1,1%), BPI (0,5%) și toxicitatea unghială (0,5%).
Tabelul reacțiilor adverse
Tabelul 4 prezintă pe scurt reacțiile adverse la medicament care au apărut la pacienții cărora li s-a administrat Rybrevant în monoterapie.
Datele reflectă expunerea la Rybrevant formă farmaceutică cu administrare intravenoasă a 380 de pacienți cu cancer pulmonar fără celule mici, avansat local sau metastazat, după eșecul chimioterapiei pe bază de platină. Pacienților li s-a administrat amivantamab 1050 mg (pentru pacienții cu greutatea < 80 kg) sau 1400 mg (pentru pacienții cu greutatea ≥80 kg). Valoarea mediană a expunerii la amivantamab a fost de 4,1 luni (interval: 0,0 până la 39,7 luni).
Reacțiile adverse observate în timpul studiilor clinice sunt enumerate mai jos pe categorii de frecvență. Categoriile de frecvențe sunt definite după cum urmează: foarte frecvente (≥1/10); frecvente (≥1/100 și < 1/10); mai puțin frecvente (≥1/1000 și < 1/100); rare (≥1/10000 și < 1/1000); foarte rare (< 1/10000); cu frecvență necunoscută (care nu poate fi estimată din datele disponibile).
În fiecare categorie de frecvență, reacțiile adverse sunt prezentate în ordinea descrescătoare a gravității.
Tabelul 4: Reacții adverse la pacienții cărora li se administrează Rybrevant în monoterapie (N=380)
| Sisteme şi organe Reacții adverse | Categoria de frecvenţă | Toate gradele (%) | Grad 3-4 (%) |
|---|---|---|---|
| Tulburări metabolice și de nutriție | |||
| Hipoalbuminemie*(vezi pct.5.1) | Foarte frecvente | 31 | 2† |
| Scăderea poftei de mâncare | 16 | 0,5† | |
| Hipocalcemie | 10 | 0,3† | |
| Hipopotasemie | Frecvente | 9 | 2 |
| Hipomagnezemie | 8 | 0 | |
| Tulburări ale sistemului nervos | |||
| Amețeală* | Foarte frecvente | 13 | 0,3† |
| Tulburări oculare | |||
| Tulburări de vedere* | Frecvente | 3 | 0 |
| Creșterea genelor* | 1 | 0 | |
| Alte tulburări oculare* | 6 | 0 | |
| Cheratită | Mai puțin frecvente | 0,5 | 0 |
| Uveită | 0,3 | 0 | |
| Tulburări respiratorii, toracice și mediastinale | |||
| Boală pulmonară interstițială* | Frecvente | 3 | 0,5† |
| Tulburări gastro-intestinale | |||
| Diaree | Foarte frecvente | 11 | 2† |
| Stomatită* | 24 | 0,5† | |
| Greață | 23 | 0,5† | |
| Constipație | 23 | 0 | |
| Vărsături | 12 | 0,5† | |
| Durere abdominală* | Frecvente | 9 | 0,8† |
| Hemoroizi | 3,7 | 0 | |
| Tulburări hepatobiliare | |||
| Valori crescute ale alanin-aminotransferazei | Foarte frecvente | 15 | 2 |
| Valori crescute ale aspartat- aminotransferazei | 13 | 1 | |
| Valori crescute ale fosfatazei alcaline serice | 12 | 0,5† | |
| Afecțiuni cutanate și ale țesutului subcutanat | |||
| Erupție cutanată tranzitorie* | Foarte frecvente | 76 | 3† |
| Toxicitate unghială* | 47 | 2† | |
| Xerodermie* | 19 | 0 | |
| Prurit | 18 | 0 | |
| Ulcerație cutanată | Mai puțin frecvente | 0,8 | 0 |
| Necroliză epidermică toxică | 0,3 | 0,3† | |
| Tulburări musculo-scheletice și ale țesutului conjunctiv | |||
| Mialgie | Foarte frecvente | 11 | 0,3† |
| Tulburări generale și la nivelul locului de administrare | |||
| Edem* | Foarte frecvente | 26 | 0,8† |
| Fatigabilitate* | 26 | 0,8† | |
| Pirexie | 11 | 0 | |
| Leziuni, intoxicații și complicații legate de procedurile utilizate | |||
| Reacții adverse legate de perfuzie | Foarte frecvente | 67 | 2 |
* Termeni grupați
† Doar reacții de grad 3
Rybrevant în asociere cu lazertinib
În general, profilul de siguranță al Rybrevant formă farmaceutică cu administrare subcutanată a fost în concordanță cu profilul de siguranță confirmat al Rybrevant formă farmaceutică cu administrare intravenoasă, cu o incidență mai scăzută a reacțiilor adverse legate de administrare și a TEV observată în cazul formei farmaceutice cu administrare subcutanată, în comparație cu forma farmaceutică cu administrare intravenoasă
În setul de date referitor la Rybrevant (formă farmaceutică cu administrare subcutanată sau formă farmaceutică cu administrare intravenoasă) în asociere cu lazertinib (N=752), cele mai frecvente reacții adverse de toate gradele ((≥20% dintre pacienți) au fost erupții cutanate tranzitorii (87%), toxicitate la nivelul unghiilor (67%), hipoalbuminemie (48%), hepatotoxicitate (43%), stomatită
(43%), edem (42%), fatigabilitate (35%), parestezie (29%), constipație (26%), diaree (26%), xerodermie (25%), scăderea poftei de mâncare (24%), greață (24%) și prurit (23%).
S-au observat diferențe relevante din punct de vedere clinic între forma farmaceutică cu administrare subcutanată și forma farmaceutică cu administrare intravenoasă, atunci când sunt administrate în asociere cu lazertinib, în ceea ce privește reacțiile legate de administrare (63% în cazul administrării intravenoase vs. 14% în cazul administrării subcutanate) și TEV (37% în cazul administrării intravenoase vs. 11% în cazul administrării subcutanate).
Reacții adverse grave au fost raportate la 14% dintre pacienții care au primit Rybrevant formă farmaceutică cu administrare subcutanată în asociere cu lazertinib, incluzând BPI (4,2%), TEV (2,7%), hepatotoxicitate (2,1%) și fatigabilitate (1,5%). Șapte la sută dintre pacienți au întrerupt administrarea Rybrevant formă farmaceutică cu administrare subcutanată din cauza reacțiilor adverse. În cazul pacienților cărora li s-a administrat Rybrevant formă farmaceutică cu administrare subcutanată în asociere cu lazertinib, cele mai frecvente reacții adverse de orice grad (≥ 1% dintre pacienți) care au condus la întreruperea tratamentului cu Rybrevant formă farmaceutică cu administrare subcutanată au fost BPI (3,6%) și erupție cutanată tranzitorie (1,5%).
Tabelul reactiilor adverse
Reacțiile adverse ale Rybrevant (formă farmaceutică cu administrare subcutanată sau formă farmaceutică cu administrare intravenoasă) atunci când este administrat în asociere cu lazertinib sunt prezentate în Tabelul 5.
Datele de siguranță de mai jos reflectă expunerea la Rybrevant (formă farmaceutică cu administrare subcutanată sau formă farmaceutică cu administrare intravenoasă) în asociere cu lazertinib la 752 de pacienți cu NSCLC în stadiu avansat local sau metastazat, inclusiv 421 de pacienți în studiul MARIPOSA, 125 de pacienți în cohortele 1 și 6 din studiul PALOMA-2 și 206 pacienți în brațul cu administrare subcutanată din studiul PALOMA-3. Pacienții au primit tratament cu Rybrevant (formă farmaceutică cu administrare subcutanată sau formă farmaceutică cu administrare intravenoasă) până la progresia bolii sau toxicitate inacceptabilă. Durata mediană a tratamentului cu amivantamab per ansamblu, atât pentru forma farmaceutică cu administrare subcutanată, cât și pentru forma farmaceutică cu administrare intravenoasă, a fost de 9,9 luni (interval: 0,1 până la 31,4 luni). Durata mediană a tratamentului pentru formă farmaceutică cu administrare subcutanată a fost de 5,7 luni (interval: 0,1 până la 13,2 luni), în timp ce durata mediană a tratamentului pentru formă farmaceutică cu administrare intravenoasă a fost de 18,5 luni (interval: 0,2 până la 31,4 luni).
Reacțiile adverse observate în timpul studiilor clinice sunt enumerate mai jos în funcție de categoria de frecvență. Categoriile de frecvențe sunt definite după cum urmează: foarte frecvente (≥1/10); frecvente (≥1/100 și < 1/10); mai puțin frecvente (≥1/1 000 și < 1/100); rare (≥1/10000 și < 1/1000); foarte rare (< 1/10000); și cu frecvență necunoscută (care nu poate fi estimată din datele disponibile).
Tabelul 5: Reacții adverse la pacienții tratați cu Rybrevant (formă farmaceutică cu administrare subcutanată sau formă farmaceutică cu administrare intravenoasă) în cazul administrării în asociere cu lazertinib (N=752)
| Clasa de aparate, sisteme și organe Reacție adversă | Categoria de frecvență | Orice grad (%) | Grad 3-4 (%) |
|---|---|---|---|
| Tulburări metabolice și de nutriție | |||
| Hipoalbuminemie* | Foarte frecvente | 48 | 4,5 |
| Apetit alimentar scăzut | 24 | 0,8 | |
| Hipocalcemie | 19 | 1,2 | |
| Hipopotasemie | 13 | 2,7 | |
| Hipomagneziemie | Frecvente | 6 | 0 |
| Tulburări ale sistemului nervos | |||
| Parestezie*,a | Foarte frecvente | 29 | 1,3 |
| Amețeală* | 12 | 0 | |
| Tulburări oculare | |||
| Alte tulburări oculare* | Foarte frecvente | 19 | 0,5 |
| Tulburări de vedere* | Frecvente | 3.6 | 0 |
| Cheratită | 1,7 | 0,3 | |
| Creștere a genelor* | 1,7 | 0 | |
| Tulburări vasculare | |||
| Tromboembolism venos | |||
| Amivantamab cu administrare intravenoasă*, b | Foarte frecvente | 37 | 11 |
| Amivantamab cu administrare subcutanată*, c | Foarte frecvente | 11 | 0,9 |
| Tulburări respiratorii, toracice și mediastinale | |||
| Boală pulmonară interstițială* | Frecvente | 3,6 | 1,7 |
| Tulburări gastro-intestinale | |||
| Stomatită* | Foarte frecvente | 43 | 2,0 |
| Constipație | 26 | 0 | |
| Diaree | 26 | 1,7 | |
| Greață | 24 | 0,8 | |
| Vărsături | 15 | 0,5 | |
| Dureri abdominale* | 10 | 0,1 | |
| Hemoroizi | Frecvente | 8 | 0,1 |
| Tulburări hepatobiliare | |||
| Hepatotoxicitate * | Foarte frecvente | 43 | 7 |
| Afecțiuni cutanate și ale țesutului subcutanat | |||
| Erupție cutanată tranzitorie* | Foarte frecvente | 87 | 23 |
| Toxicitate la nivelul unghiilor* | 67 | 8 | |
| Piele uscată* | 25 | 0,7 | |
| Prurit | 23 | 0,3 | |
| Ulcerație cutanată | Frecvente | 3,9 | 0,5 |
| Sindromul de eritrodisestezie palmo-plantară | 3,9 | 0,1 | |
| Urticarie | 1,6 | 0 | |
| Tulburări musculo-scheletice și ale țesutului conjunctiv | |||
| Mialgie | Foarte frecvente | 15 | 0,5 |
| Spasme musculare | 13 | 0,4 | |
| Tulburări generale și la nivelul locului de administrare | |||
| Edem* | Foarte frecvente | 42 | 2,7 |
| Fatigabilitate* | 35 | 3,5 | |
| Pirexie | 11 | 0 | |
| Reacții la locul de injectare*, c, d | Frecvente | 8 | 0 |
| Leziuni, intoxicații și complicații legate de proceduri | |||
| Reacții adverse legată de perfuzie/administrare | |||
| Amivantamab cu administrare intravenoasăb, e | Foarte frecvente | 63 | 6 |
| Amivantamab cu administrare subcutanatăc, f | Foarte frecvente | 14 | 0,3 |
* Termeni grupați. a
Se aplică doar pentru lazertinib. b
Frecvență bazată doar pe studiul cu amivantamab cu administrare intravenoasă (MARIPOSA [N=421]). c
Frecvență bazată doar pe studiile cu amivantamab cu administrare subcutanată (cohortele 1 și 6 din studiul PALOMA- 2 [N=125] și brațul cu administrare subcutanată din studiul PALOMA-3 [N=206]). d
Reacțiile adverse la locul de injectare sunt semne și simptome locale asociate cu modul de administrare subcutanat. e
Reacțiile adverse legate de perfuzie sunt semne și simptome sistemice asociate cu administrarea intravenoasă a perfuziei cu amivantamab. f
Reacțiile adverse legate de administrare sunt semne și simptome sistemice asociate cu administrarea de amivantamab subcutanat.
Descrierea reacțiilor adverse selectate
Reacții adverse legate de administrare
În total, reacțiile adverse legate de administrare au apărut la 14% dintre pacienții tratați cu Rybrevant formă farmaceutică cu administrare subcutanată în asociere cu lazertinib. În studiul PALOMA-3, reacțiile adverse legate de administrare au fost raportate la 13% dintre pacienții tratați cu Rybrevant formă farmaceutică cu administrare subcutanată în asociere cu lazertinib, comparativ cu 66% dintre pacienții tratați cu Rybrevant formă farmaceutică cu administrare intravenoasă în asociere cu lazertinib. Cele mai frecvente semne și simptome ale reacțiilor adverse legate de administrare includ dispnee, înroșire a feței, febră, frisoane, greață și disconfort toracic. Timpul median până la apariția primelor reacții adverse legate de administrare a fost de 2,1 ore (interval: 0,0 până la 176,5 ore). Majoritatea reacțiilor adverse legate de administrare (98%) au fost de gradul 1 sau 2 din punct de vedere al severității.
Reacții adverse la locul de injectare
În total, au apărut reacții adverse la locul de injectare la 8% dintre pacienții tratați cu Rybrevant formă farmaceutică cu administrare subcutanată în asociere cu lazertinib. Toate reacțiile adverse la locul injectării au fost de gradul 1 sau 2 din punct de vedere al severității. Cel mai frecvent simptom al reacțiilor adverse la locul de injectare a fost eritemul.
Boală pulmonară interstițială
Boala pulmonară interstițială (BPI) sau reacțiile adverse similare BPI au fost raportate la utilizarea amivantamab, precum și la administrarea altor inhibitori ai EGFR. BPI a fost raportată la 3,6% dintre pacienții tratați cu Rybrevant (formă farmaceutică cu administrare subcutanată sau formă farmaceutică cu administrare intravenoasă) în asociere cu lazertinib, inclusiv 2 (0,3%) pacienți care au prezentat o reacție letală. Pacienții cu istoric medical de BPI, inclusiv BPI indusă medicamentos sau pneumonită de iradiere, au fost excluși din studiile clinice PALOMA-2 și PALOMA-3.
Evenimente tromboembolice venoase (TEV) asociate cu administrarea în asociere cu lazertinib
Evenimentele TEV, inclusiv tromboza venoasă profundă (TVP) și embolia pulmonară (EP), au fost raportate la 11% dintre pacienții care au primit Rybrevant formă farmaceutică cu administrare subcutanată în asociere cu lazertinib în studiile clinice PALOMA-2 și PALOMA-3. Majoritatea evenimentelor au fost de gradul 1 sau 2, evenimente de gradul 3 survenind la 3 (0,9%) pacienți. În plus, 269 (81%) dintre acești 331 de pacienți care au primit Rybrevant formă farmaceutică cu administrare subcutanată au luat tratament anticoagulant profilactic, cu un anticoagulant oral direct sau heparină cu greutate moleculară mică, în primele patru luni de tratament în cadrul studiului. În studiul clinic PALOMA-3, incidența evenimentelor TEV a fost de 9% în cazul pacienților tratați cu Rybrevant formă farmaceutică cu administrare subcutanată în asociere cu lazertinib, în comparație cu 13% în cazul pacienților tratați cu Rybrevant formă farmaceutică cu administrare intravenoasă în asociere cu lazertinib, cu rate similare de utilizare a tratamentului anticoagulant profilactic în cadrul ambelor brațe de tratament (80% în brațul cu administrare subcutanată vs. 81% în brațul cu administrare intravenoasă). Pentru pacienții care nu au primit tratament anticoagulant profilactic, incidența generală a TEV a fost de 17% pentru pacienții tratați cu Rybrevant formă farmaceutică cu administrare subcutanată în asociere cu lazertinib, cu majoritatea evenimentelor TEV raportate ca fiind de gradul 1- 2 și evenimente TEV grave raportate la 4,8% dintre acești pacienți, în comparație cu o incidență generală de 23% pentru pacienții tratați cu Rybrevant formă farmaceutică cu administrare intravenoasă în asociere cu lazertinib, cu evenimente TEV de gradul 3 raportate la 10% și evenimente TEV grave raportate la 8% dintre acești pacienți
Reacții cutanate și unghiale
Erupțiile cutanate tranzitorii (inclusiv dermatita acneiformă), pruritul și pielea uscată au apărut la pacienții tratați cu Rybrevant (formă farmaceutică cu administrare subcutanată sau formă farmaceutică cu administrare intravenoasă) în asociere cu lazertinib. Erupțiile cutanate tranzitorii au apărut la 87% dintre pacienți, ducând la întreruperea tratamentului cu Rybrevant la 0,7% dintre pacienți. Majoritatea cazurilor au fost de gradul 1 sau 2, cu reacții adverse de gradul 3 și 4 apărute la 23% și, respectiv, 0,1% dintre pacienți.
A fost realizat un studiu de fază 2 la pacienții tratați cu Rybrevant în asociere cu lazertinib pentru a evalua utilizarea terapiei profilactice cu un antibiotic cu utilizare orală, un antibiotic administrat topic la nivelul scalpului, o cremă hidratantă pe față și pe întreg corpul (cu excepția scalpului) și un antiseptic pe mâini și picioare (vezi pct. 4.2 și 4.4). S-a demonstrat o reducere a incidenței evenimentelor adverse dermatologice de grad ≥2 în primele 12 săptămâni de tratament, comparativ cu tratamentul dermatologic standard utilizat în practica clinică (38,6% față de 76,5%, p<0,0001). În plus, s-a observat o reducere a evenimentelor adverse de grad ≥2 care au afectat scalpul în primele 12 săptămâni de tratament (8,6% față de 29,4%), împreună cu o incidență mai mică a reducerilor de doză (7,1% față de 19,1%), a întreruperilor administrării (15,7% față de 33,8%) și a opririi tratamentului (1,4% față de 4,4%) din cauza evenimentelor adverse dermatologice.
Tulburări oculare
Tulburări oculare, inclusiv cheratită (1,7%), au apărut la pacienții tratați cu Rybrevant (formă farmaceutică cu administrare subcutanată sau formă farmaceutică cu administrare intravenoasă). Alte reacții adverse raportate au inclus creșterea genelor, afectarea vederii și alte tulburări oculare.
Categorii speciale de populație
Vârstnici
Există informații clinice limitate privind administrarea amivantamab la pacienții cu vârstă ≥75 ani (vezi pct. 5.1). În general, nu s-au observat diferențe în ceea ce privește siguranța administrării la pacienții cu vârstă ≥65 ani față de pacienții cu vârstă < 65 ani.
Raportarea reacțiilor adverse suspectate
Raportarea reacțiilor adverse suspectate după autorizarea medicamentului este importantă. Acest lucru permite monitorizarea continuă a raportului beneficiu/risc al medicamentului. Profesioniștii din domeniul sănătății sunt rugați să raporteze orice suspiciune de reacție adversă prin intermediul sistemului național de raportare, așa cum este menționat în Anexa V.
4.9 Supradozaj
Nu există informații privind supradozajul cu Rybrevant formă farmaceutică cu administrare subcutanată și niciun antidot specific cunoscut pentru supradozaj. În cazul unui supradozaj, tratamentul cu Rybrevant trebuie întrerupt, pacientul trebuie monitorizat pentru orice semne sau simptome de evenimente adverse și trebuie instituite imediat măsuri generale adecvate de asistență până la diminuarea sau remisiunea toxicității clinice.
5. PROPRIETĂȚI FARMACOLOGICE
5.1 Proprietăți farmacodinamice
Grupa farmacoterapeutică: Anticorpi monoclonali și conjugați anticorp-medicament, codul ATC: L01FX18.
Rybrevant formă farmaceutică cu administrare subcutanată conține hialuronidază umană recombinantă (rHuPH20). rHuPH20 acționează local și temporar pentru a degrada hialuronanul ((HA), un glicozaminoglican natural care se găsește în întregul organism) din matricea extracelulară a spațiului subcutanat, prin scindarea legăturii dintre cele două glucide (N-acetilglucozamină și acid glucuronic) care alcătuiesc HA.
Mecanism de acțiune
Amivantamab este un anticorp bispecific EGFR-TME cu conținut scăzut de fucoză, complet uman, de tip IgG1, cu activitate imunitară mediată celular, care vizează tumorile cu mutații activatoare de EGFR, cum ar fi delețiile în Exonul 19, substituția în Exonul 21 L858R și mutațiile de inserție în Exonul 20. Amivantamab se leagă de domeniile extracelulare ale EGFR și TME.
Amivantamab întrerupe funcțiile de semnalizare ale EGFR și TME prin blocarea legării ligandului și intensificarea degradării EGFR și a TME, prevenind astfel creșterea și progresia tumorii. Prezența EGFR și MET pe suprafața celulelor tumorale permite, de asemenea, țintirea acestor celule pentru distrugere de către celulele efectoare imune, cum ar fi celulele natural killer și macrofagele, prin citotoxicitate dependentă de anticorpi mediată celular (CDAC) și, respectiv, mecanisme de trogocitoză.
Efecte farmacodinamice
După prima doză completă de Rybrevant formă farmaceutică cu administrare subcutanată, concentrațiile serice medii de EGFR și TME au scăzut substanțial și s-au menținut suprimate pe parcursul tratamentului pentru toate dozele studiate.
Albumină
Rybrevant formă farmaceutică cu administrare subcutanată a scăzut concentrația serică de albumină, un efect farmacodinamic al inhibării TME, de obicei în primele 8 săptămâni (vezi pct. 4.8); ulterior, concentrația de albumină s-a stabilizat pentru restul tratamentului cu amivantamab.
Experiența clinică cu Rybrevant formă farmaceutică cu administrare subcutanată
Eficacitatea Rybrevant formă farmaceutică cu administrare subcutanată la pacienții cu NSCLC avansat local sau metastazat, cu mutație EGFR, se bazează pe obținerea unei expuneri farmacocinetice noninferioare la amivantamab cu administrare intravenoasă în studiul de non-inferioritate PALOMA-3 (vezi pct. 5.2). Studiul a demonstrat eficacitatea non-inferioară a amivantamab cu administrare subcutanată comparativ cu amivantamab cu administrare intravenoasă, în asociere cu lazertinib la pacienții cu NSCLC avansat local sau metastazat, cu mutație EGFR, a căror boală a progresat în timpul sau după tratamentul cu osimertinib și chimioterapie pe bază de platină.
Experiența clinică cu Rybrevant formă farmaceutică cu administrare intravenoasă
Cancer pulmonar fără celule mici (NSCLC) netratat anterior, cu deleții în Exonul 19 al EGFR sau cu mutații de substituție în Exonul 21 L858R (MARIPOSA)
NSC3003 (MARIPOSA) este un studiu de fază 3, multicentric, controlat activ, în regim deschis, randomizat, de evaluare a eficacității și a siguranței Rybrevant formă farmaceutică cu administrare intravenoasă în asociere cu lazertinib comparativ cu osimertinib în monoterapie ca tratament de primă linie la pacienții cu NSCLC cu mutații EGFR, avansat local sau metastatic, care nu răspunde la terapia curativă. Trebuia ca probele pacienților să aibă una dintre cele două mutații comune din EGFR (deleție a Exonului 19 sau mutație de substituție L858R a Exonului 21), identificată prin testare la nivel local. Probele de țesut tumoral (94%) și/sau de plasmă (6%) prelevate de la toți pacienții au fost testate la nivel local pentru a determina statusul deleției Exonului 19 al EGFR și/sau al mutației de substituție L858R a Exonului 21, utilizând reacția de polimerizare în lanț (polymerase chain reaction, PCR) la 65% dintre pacienți și secvențierea de generație următoare (NGS) la 35% dintre pacienți.
În total, 1074 de pacienți au fost randomizați (în raport de 2:2:1) pentru a li se administra Rybrevant formă farmaceutică cu administrare intravenoasă în asociere cu lazertinib, osimertinib în monoterapie sau lazertinib în monoterapie până la progresia bolii sau până la toxicitate inacceptabilă. Rybrevant formă farmaceutică cu administrare intravenoasă a fost administrat intravenos în doze de 1050 mg (la pacienți cu greutatea < 80 kg) sau de 1400 mg (la pacienți cu greutatea ≥80 kg), o dată pe săptămână timp de 4 săptămâni, și ulterior, începând cu săptămâna 5, la fiecare 2 săptămâni. Lazertinib a fost administrat în doze de 240 mg oral o dată pe zi. Osimertinib a fost administrat în doze de 80 mg oral o dată pe zi. Randomizarea a fost stratificată în funcție de tipul mutației din EGFR (deleție a Exonului 19 sau Exonul 21 L858R), rasă (asiatică sau non-asiatică) și antecedente de metastaze cerebrale (da sau nu).
Caracteristicile demografice și ale bolii la momentul inițial au fost echilibrate între grupurile de tratament. Mediana vârstei a fost de 63 (interval: 25-88) de ani, 45% dintre pacienți având vârsta ≥65 de ani; 62% au fost femei; 59% au fost asiatici și 38% caucazieni. Statusul de performanță la momentul inițial al Grupului Estic pentru Cooperare în Oncologie (Eastern Cooperative Oncology Group, ECOG) a fost 0 (34%) sau 1 (66%); 69% nu fumaseră niciodată; 41% aveau metastaze cerebrale anterioare, iar 90% aveau cancer în stadiul IV la diagnosticul inițial. În ceea ce privește statusul mutațiilor la nivelul EGFR, 60% erau deleții ale Exonului 19 și 40% erau mutații de substituție L858R ale Exonului 21.
Rybrevant formă farmaceutică cu administrare intravenoasă în asociere cu lazertinib a demonstrat o îmbunătățire semnificativă statistic a supraviețuirii fără progresia bolii (SFP) pe baza evaluării BICR.
Analiza finală privind SG a demonstrat o îmbunătățire semnificativă din punct de vedere statistic a SG în cazul tratamentului cu Rybrevant în asociere cu lazertinib în comparație cu tratamentul cu osimertinib (vezi Tabelul 6 și Figura 2).
Tabelul 6: Rezultate privind eficacitatea în studiul MARIPOSA
| Rybrevant formă farmaceutică cu administrare intravenoasă + lazertinib (N=429) | Osimertinib (N=429) | |
| Supraviețuire fără progresia bolii (SFP)a | ||
|---|---|---|
| Număr de evenimente | 192 (45%) | 252 (59%) |
| Mediană, luni (IÎ 95%) | 2,7 (19,1, 27,7) | 16,6 (14,8, 18,5) |
| Risc relativ (IÎ 95%); valoarea p | 0,70 (0,58, 0,85); p=0,0002 | |
| Supraviețuire globală (SG) | ||
| Număr de evenimente | 173 (40%) | 217 (51%) |
| Mediană, luni (IÎ 95%) | NE (42,9, NE) | 36,7 (33,4, 41,0) |
| Risc relativ (IÎ 95%); valoarea p | 0,75 (0,61, 0,92); p=0,0048 | |
| Rata răspunsului obiectiv (RRO)a,b | ||
| RRO % (IÎ 95%) | 80% (76%, 84%) | 77% (72%, 81%) |
| Durata răspunsului (DR)a,b | ||
| Mediană, luni (IÎ 95%) | 25,8 (20,3, 33,9) | 18,1 (14,8, 20,1) |
BICR = analiză centrală independentă în regim orb; IÎ = interval de încredere; NE = nu se poate estima.
Rezultatele privind SFP se referă la data centralizării datelor 11 august 2023, cu o perioadă mediană de urmărire de 22,0 luni. Rezultatele privind RRO și DR se referă la data centralizării datelor 13 mai 2024, cu o perioadă mediană de urmărire de 31,3 luni. Rezultatele privind SG se referă la data centralizării datelor 4 decembrie 2024, cu o perioadă mediană de urmărire de 37,8 luni. a
BICR pe baza criteriilor RECIST v1.1. b
Pe baza respondenților confirmați.
Figura 1: Curba Kaplan-Meier a SFP la pacienți cu NSCLC netratați anterior, obținută în urma evaluării BICR
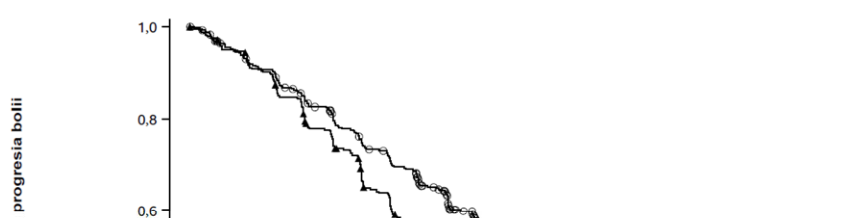


Figura 2: Curba Kaplan-Meier a SG la pacienți cu NSCLC netratați anterior


În studiul MARIPOSA, RRO intracranian și DR pe baza evaluării BICR au fost criterii finale de evaluare pre-specificate. În subsetul de pacienți cu leziuni intracraniene la momentul inițial, asocierea
Rybrevant formă farmaceutică cu administrare intravenoasă și lazertinib a demonstrat un RRO intracranian similar cu cel al elementului de control. Conform protocolului, toți pacienții din studiul MARIPOSA au fost supuși unei serii de examinări IRM pentru a evalua răspunsul intracranian și durata. Rezultatele sunt prezentate pe scurt în Tabelul 7.
Tabelul 7: RRO intracranian și DR pe baza evaluării BICR la subiecții cu leziuni intracraniene la momentul inițial - MARIPOSA
| Rybrevant formă farmaceutică cu administrare intravenoasă + lazertinib (N=180) | Osimertinib (N=186) | |
| Evaluarea răspunsului tumoral intracranian | ||
|---|---|---|
| RRO intracranian (RC+RP), % (IÎ 95%) | 78% (71%, 84%) | 77% (71%, 83%) |
| Răspuns complet | 64% | 59% |
| DR intracranian | ||
| Număr de respondenți | 140 | 144 |
| Mediană, luni (IÎ 95%) | 35,0 (20,4, NE) | 25,1(22,1, 31,2) |
IÎ = interval de încredere
NE = nu se poate estima
Rezultatele privind RRO intracranian și DR se referă la data centralizării datelor 4 decembrie 2024, cu o perioadă mediană de urmărire de 37,8 luni.
Cancer pulmonar fără celule mici (NSCLC), tratat anterior, cu mutații ale inserției Exon 20 (CHRYSALIS)
CHRYSALIS este un studiu multicentric, în regim deschis, cu mai multe cohorte, efectuat pentru a evalua siguranța și eficacitatea Rybrevant formă farmaceutică cu administrare intravenoasă la pacienții cu NSCLC avansat local sau metastazat. Eficacitatea a fost evaluată la 114 pacienți cu NSCLC avansat local sau metastazat, care prezentau mutații de inserție Exon 20 EGFR, a căror boală progresase în timpul sau după chimioterapia pe bază de platină și care au avut o perioadă mediană de urmărire de 12,5 luni. Eșantioane de țesut tumoral (93%) și/sau plasmatic (10%) pentru toți pacienții au fost testate la nivel local pentru a determina statusul mutației de inserție Exon 20 EGFR utilizând secvențierea de generație următoare (NGS) la 46% dintre pacienți și/sau reacția în lanț a polimerazei (PCR) la 41% dintre pacienți; pentru 4% dintre pacienți, metodele de testare nu au fost specificate. Pacienții care au prezentat în ultimii 2 ani metastaze cerebrale netratate sau cu antecedente de boli pulmonare interstițiale (BPI) care necesită tratament cu steroizi cu acțiune prelungită sau alți agenți imunosupresori nu au fost eligibili pentru studiu. Rybrevant formă farmaceutică cu administrare intravenoasă a fost administrat intravenos în doză de 1050 mg la pacienți cu greutatea < 80 kg sau 1400 mg la pacienți cu greutatea ≥80 kg, o dată pe săptămână, timp de 4 săptămâni, apoi la fiecare 2 săptămâni, începând cu săptămâna 5, până la dispariția beneficiului clinic sau a toxicității inacceptabile. Criteriul final principal de evaluare a eficacității a fost rata de răspuns global (RRG) evaluată de investigator, definită ca răspuns complet (RC) sau răspuns parțial (RP) confirmat pe baza RECIST v1.1. În plus, criteriul de evaluare final principal a fost evaluat prin intermediul unei evaluări centrale independente în regim orb (BICR). Criteriile finale secundare de evaluare a eficacității au inclus durata răspunsului (DR).
Vârsta mediană a fost de 62 ani (interval: 36–84) ani, cu 41% dintre pacienți cu vârsta ≥65 ani; 61% au fost femei; și 52% au fost asiatici și 37% au fost caucazieni. Numărul median de tratamente anterioare a fost de 2 (interval: 1 până la 7 terapii). La momentul inițial, 29% din pacienți aveau scorul de performanță ECOG de 0 și 70% aveau scorul de performanță ECOG de 1; 57% nu au fumat niciodată; 100% aveau cancer în stadiul IV; iar 25% aveau tratament anterior pentru metastaze cerebrale. Inserțiile din Exon 20 au fost observate la 8 reziduuri diferite; cele mai frecvente reziduuri au fost A767 (22%), S768 (16%), D770 (12%) și N771 (11%).
Rezultatele cu privire la eficacitate sunt prezentate pe scurt în Tabelul 8.
Tabelul 8: Rezultatele eficacității în studiul CHRYSALIS
| Evaluare Investigator (N=114) | |
| Rata de răspuns globala, b(IÎ 95%) | 37% (28%, 46%) |
| Răspuns complet | 0% |
| Răspuns parțial | 37% |
| Durata răspunsului | |
|---|---|
| Valoarea medianăc(IÎ 95%), luni | 12,5 (6,5; 16,1) |
| Pacienți cu DR ≥6 luni | 64% |
IÎ = Interval de încredere a
Răspuns confirmat b
Rezultatele RRG și DR obținute în urma evaluării efectuate de investigator au fost similare cu cele raportate în urma evaluării BICR; valorile RRG obținute de BICR au fost 43% (34%, 53%), cu 3% rata RC și 40% rata RP, DR mediană obținută de BICR a fost 10,8 luni (IÎ 95%: 6,9, 15,0) iar pacienții cu DR ≥6 luni conform evaluării BICR au reprezentat 55%. c
Pe baza estimării Kaplan-Meier.
Activitatea antitumorală a fost observată în toate cazurile purtătoare de mutație.
Imunogenitatea
Anticorpii anti-medicament (AAM) au fost detectați mai puțin frecvent după tratamentul cu Rybrevant formă farmaceutică cu administrare subcutanată. Nu a fost observată nicio dovadă a impactului AAM asupra farmacocineticii, eficacității sau siguranței. Dintre cei 389 de participanți care au primit Rybrevant formă farmaceutică cu administrare subcutanată în monoterapie sau ca parte a unei asocieri terapeutice, 37 de participanți (10%) au fost testați pozitivi pentru anticorpi anti-rHuPH20 apăruți în timpul tratamentului. Imunogenitatea la rHuPH20 observată la acești participanți nu a avut impact asupra farmacocineticii amivantamab.
Vârstnici
Nu s-au observat diferențe la modul general în ceea ce privește eficacitatea la pacienții cu vârstă ≥65 ani față de pacienții cu vârstă < 65 ani.
Copii și adolescenți
Agenția Europeană pentru Medicamente a acordat o derogare de la obligația de depunere a rezultatelor studiilor efectuate cu Rybrevant la toate subgrupele de copii și adolescenți în tratamentul NSCLC (vezi pct. 4.2 pentru informații privind utilizarea la copii și adolescenți).
5.2 Proprietăți farmacocinetice
Absorbție
În urma administrării subcutanate, media geometrică (%CV) a biodisponibilității amivantamabului este de 66,6% (14,9%), cu un timp median până la atingerea concentrației maxime de 3 zile, pe baza estimărilor parametrilor farmacocinetici individuali ai amivantamabului pentru participanții cărora li sa administrat subcutanat, în cadrul analizei farmacocinetice populaționale.
În cazul schemei de administrare subcutanată a dozei o dată la 2 săptămâni, media geometrică (%CV) a valorilor maxime ale concentrației plasmatice minime de amivantamab după cea de-a 4-a doză săptămânală a fost de 335 µg/ml (32,7%). ASC1 săptămână medie a crescut de 3,5 ori de la prima doză până în ziua 1 a ciclului 2. Valoarea maximă a concentrației plasmatice minime de amivantamab după administrarea subcutanată în monoterapie și în asociere cu lazertinib este observată, de obicei, la sfârșitul schemei de administrare săptămânale (ciclul 2 ziua 1). Concentrația plasmatică la starea de echilibru a amivantamab este atinsă până în săptămâna 13. Media geometrică (%CV) a concentrațiilor plasmatice minime la starea de echilibru a amivantamab în ciclul 4 ziua 1 a fost de 206 µg/ml (39,1%).
Tabelul 9 prezintă media geometrică observată (%CV) a valorilor maxime ale concentrațiilor plasmatice minime (Ctrough din ciclul 2 ziua 1) și aria de sub curba concentrație - timp în ciclul 2
(ASCziua 1-15) după dozele recomandate de amivantamab cu administrare subcutanată și intravenoasă la pacienții cu NSCLC. Acești parametri farmacocinetici au stat la baza demonstrației de noninferioritate care susține trecerea de la administrarea intravenoasă la cea subcutanată.
Tabelul 9: Rezumatul parametrilor farmacocinetici plasmatici ai amivantamab la pacienții cu NSCLC (Studiul PALOMA-3)
| Parametru | Rybrevant formă farmaceutică cu administrare subcutanată 1600 mg (2240 mg pentru greutate corporală ≥80 kg) | Rybrevant formă farmaceutică cu administrare intravenoasă 1050 mg (1400 mg pentru greutate corporală ≥80 kg) |
|---|---|---|
| Media geometrică (%CV) | ||
| Ctrough din ciclul 2 ziua 1 (µg/ml) | 335 (32,7%) | 293 (31,7%) |
| AUC(Ziua 1-15) în ciclul 2 (µg/ml) | 135861 (30,7%) | 131704 (24,0%) |
Distribuție
Pe baza estimărilor parametrilor farmacocinetici individuali ai amivantamab la pacienții cărora li s-a administrat forma farmaceutică cu administrare subcutanată, din analiza farmacocinetică populațională, valoarea mediei geometrice (CV%) a volumului total de distribuție al amivantamab administrat subcutanat este 5,69 l (23,8%).
Eliminare
Pe baza estimărilor parametrilor farmacocinetici individuali ai amivantamab la pacienții cărora li s-a administrat forma farmaceutică cu administrare subcutanată, din analiza farmacocinetică populațională, valoarea mediei geometrice (CV%) a clearance-ului liniar (CL) și a timpului de înjumătățire plasmatică asociat cu clearance-ul liniar este de 0,224 l/zi (26,0%) și, respectiv, 18,8 zile (34,3%).
Categorii speciale de populație
Vârstnici
Nu s-au observat diferențe semnificative din punct de vedere clinic în ceea ce privește farmacocinetica amivantamab în funcție de vârstă (21-88 de ani).
Insuficiență renală
Nu s-a observat niciun efect semnificativ din punct de vedere clinic asupra farmacocineticii amivantamab la pacienții cu insuficiență renală ușoară (60 ≤ clearance-ul creatininei [ClCr] < 90 ml/min), moderată (29 ≤ ClCr < 60 ml/min) sau severă (15 ≤ CrCl < 29 ml/min). Datele referitoare la pacienții cu insuficiență renală severă sunt limitate (n=1), dar nu există dovezi care să sugereze necesitatea ajustării dozei la acești pacienți. Nu se cunoaște efectul bolii renale în stadiu terminal (ClCr < 15 ml/min) asupra farmacocineticii amivantamabului.
Insuficiență hepatică
Este puțin probabil ca modificările funcției hepatice să aibă vreun efect asupra eliminării amivantamabului, deoarece moleculele pe bază de IgG1, cum este amivantamabul, nu sunt metabolizate pe cale hepatică.
Nu s-a observat niciun efect semnificativ din punct de vedere clinic asupra farmacocineticii amivantamab pe baza insuficienței hepatice ușoare [(bilirubina totală ≤LSVN și AST > LSVN) sau (LSVN < bilirubina totală ≤1,5 x LSVN)] sau moderate (1,5×LSVN < bilirubina totală ≤3×LSVN și orice AST). Datele referitoare la pacienții cu insuficiență hepatică moderată sunt limitate (n=1), dar nu există dovezi care să sugereze necesitatea ajustării dozei la acești pacienți. Nu se cunoaște efectul insuficienței hepatice severe (bilirubină totală > 3 ori LSVN) asupra farmacocineticii amivantamabului.
Copii și adolescenți
Nu s-a studiat farmacocinetica amivantamabului la copii și adolescenți.
5.3 Date preclinice de siguranță
Datele non-clinice nu au evidențiat niciun risc special pentru om pe baza studiilor convenționale farmacologice privind toxicitatea după doze repetate.
Carcinogenitate și mutagenitate
Nu au fost efectuate studii la animale pentru a stabili potențialul carcinogen al amivantamabului. Studiile de genotoxicitate și carcinogenitate de rutină nu sunt, în general, aplicabile produselor farmaceutice biologice, deoarece proteinele mari nu pot difuza în celule și nu pot interacționa cu ADN-ul sau materialul cromozomial.
Toxicitate asupra funcției de reproducere
Nu au fost efectuate studii la animale pentru a evalua efectele asupra reproducerii și dezvoltării fetale; cu toate acestea, pe baza mecanismului său de acțiune, amivantamabul poate cauza leziuni fetale sau anomalii de dezvoltare. Așa cum este raportat în literatura de specialitate, reducerea, eliminarea sau întreruperea semnalizării EGFR embrio-fetale sau materne pot preveni implantarea, pot cauza pierderea embrionului fetal în timpul diferitelor etape ale gestației (prin efecte asupra dezvoltării placentare), pot cauza anomalii de dezvoltare la nivelul mai multor organe sau moartea precoce la fetușii supraviețuitori. În mod similar, inactivarea TME sau a factorului său de creștere hepatocit ligand (HGF) a fost embrionar letal datorită defectelor severe în dezvoltarea placentară, iar fetușii au prezentat defecte în dezvoltarea musculară la nivelul mai multor organe. Se cunoaște că IgG1 uman traversează placenta; prin urmare, amivantamabul are potențial de a fi transmis de la mamă la fătul în dezvoltare.
6. PROPRIETĂȚI FARMACEUTICE
6.1 Lista excipienților
Hialuronidază umană recombinantă (rHuPH20)
Acid etilendiaminotetraacetic (EDTA)
Acid acetic glacial
L-metionină
Polisorbat 80 (E433)
Acetat de sodiu trihidrat
Sucroză
Apă pentru preparate injectabile
6.2 Incompatibilități
Acest medicament nu trebuie amestecat cu alte medicamente, cu excepția celor menționate la pct. 6.6.
6.3 Perioada de valabilitate
Flacon nedeschis
2 ani
Soluție pregătită în seringă
Stabilitatea chimică și fizică în uz a fost demonstrată pentru o perioadă de până la 24 de ore la temperaturi de 2°C până la 8°C urmată de o perioadă de până la 24 ore la temperaturi de 15°C până la 30°C. Din punct de vedere microbiologic, cu excepția situației în care metoda de preparare a dozei exclude riscul de contaminare microbiană, produsul trebuie administrat imediat. În cazul în care produsul nu se administrează imediat, responsabilitatea în ceea ce privește timpul și condițiile de păstrare în uz revine utilizatorului.
6.4 Precauții speciale pentru păstrare
A se păstra la frigider (2°C la 8°C).
A nu se congela.
A se păstra în ambalajul original, pentru a fi protejat de lumină.
Pentru condițiile de păstrare după pregătirea seringii, vezi pct. 6.3.
6.5 Natura și conținutul ambalajului
10 ml de soluție într-un flacon din sticlă de tip 1 cu capac din elastomer și capsă din aluminiu cu cap detașabil, conținând amivantamab 1600 mg. Pachet cu 1 flacon.
14 ml de soluție într-un flacon din sticlă de tip 1 cu capac din elastomer și capsă din aluminiu cu cap detașabil, conținând amivantamab 2240 mg. Pachet cu 1 flacon.
6.6 Precauții speciale pentru eliminarea reziduurilor și alte instrucțiuni de manipulare
Rybrevant formă farmaceutică cu administrare subcutanată este de unică folosință și este gata de utilizare.
A se prepara soluția injectabilă utilizând o tehnică aseptică după cum urmează:
Preparare
- Se stabilește doza necesară și flaconul adecvat de Rybrevant formă farmaceutică cu administrare subcutanată necesar în funcție de greutatea inițială a pacientului (vezi pct. 4.2).
- Pacienților cu greutate corporală < 80 kg li se administrează 1600 mg, iar pacienților cu greutate corporală ≥80 kg, li se administrează 2240 mg o dată pe săptămână din săptămâna 1 până în săptămâna 4 și, apoi, la fiecare 2 săptămâni începând cu săptămâna 5.
- Se scoate flaconul corespunzător de Rybrevant formă farmaceutică cu administrare subcutanată din frigider (2°C până la 8°C).
- Se verifică dacă soluția Rybrevant este incoloră până la galben deschis. A nu se utiliza în caz de schimbare a culorii sau dacă sunt prezente particule opace sau alte particule străine.
- Se lasă Rybrevant formă farmaceutică cu administrare subcutanată să ajungă la temperatura camerei (15°C până la 30°C) timp de cel puțin 15 minute. A nu se încălzi Rybrevant formă farmaceutică cu administrare subcutanată prin nicio altă metodă. A nu se agita.
- Se extrage volumul necesar de Rybrevant formă farmaceutică cu administrare subcutanată pentru injecție, din flacon, într-o seringă de mărime corespunzătoare, folosind un ac de transfer. Seringile de dimensiuni mai mici necesită mai puțină forță în timpul preparării și administrării.
- Rybrevant formă farmaceutică cu administrare subcutanată este compatibil cu ace de injectare din oțel inoxidabil, seringi din polipropilenă și policarbonat și seturi de perfuzie subcutanată din polietilenă, poliuretan și policlorură de vinil. O soluție de clorură de sodiu 9 mg/ml (0,9%) poate fi, de asemenea, utilizată pentru lavajul setului de perfuzie, dacă este necesar.
- Se înlocuiește acul de transfer cu accesoriile corespunzătoare pentru transport sau administrare. Se recomandă utilizarea unui ac cu dimensiunea între 21G și 23G sau a unui set de perfuzie pentru a asigura ușurința administrării.
Păstrarea seringii pregătite
Conținutul seringii pregătite trebuie administrat imediat. În cazul în care administrarea imediată nu este posibilă, seringa pregătită se păstrează la frigider, la o temperatură cuprinsă între 2°C și 8°C, o perioadă de până la 24 de ore, apoi la temperatura camerei, între 15°C și 30°C, o perioadă de până la 24 de ore. Seringa pregătită trebuie aruncată dacă se păstrează mai mult de 24 de ore la frigider sau mai mult de 24 de ore la temperatura camerei. Dacă este păstrată în frigider, temperatura soluției trebuie să revină la temperatura camerei înainte de administrare.
Eliminare
Acest medicament este de unică folosință. Orice medicament neutilizat sau deșeu trebuie eliminat în conformitate cu reglementările locale.
7. DEȚINĂTORUL AUTORIZAȚIEI DE PUNERE PE PIAȚĂ
Janssen-Cilag International NV
Turnhoutseweg 30
B-2340 Beerse
Belgia
8. NUMĂRUL(ELE) AUTORIZAȚIEI DE PUNERE PE PIAȚĂ
EU/1/21/1594/002
EU/1/21/1594/003
9. DATA PRIMEI AUTORIZĂRI SAU A REÎNNOIRII AUTORIZAȚIEI
Data primei autorizări: 09 decembrie 2021
Data ultimei reînnoiri a autorizației: 11 septembrie 2023
10. DATA REVIZUIRII TEXTULUI
Informații detaliate privind acest medicament sunt disponibile pe site-ul Agenției Europene pentru Medicamente https://www.ema.europa.eu.