LAZCLUZE 240 mg
Rezumatul caracteristicilor produsului (RCP)
- 1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI
- 2. COMPOZIȚIA CALITATIVĂ ȘI CANTITATIVĂ
- 3. FORMA FARMACEUTICĂ
-
4. DATE CLINICE
- 4.1 Indicații terapeutice
- 4.2 Doze și mod de administrare
- 4.3 Contraindicații
- 4.4 Atenționări și precauții speciale pentru utilizare
- 4.5 Interacțiuni cu alte medicamente și alte forme de interacțiune
- 4.6 Fertilitatea, sarcina și alăptarea
- 4.7 Efecte asupra capacității de a conduce vehicule și de a folosi utilaje
- 4.8 Reacții adverse
- 4.9 Supradozaj
- 5. PROPRIETĂȚI FARMACOLOGICE
- 6. PROPRIETĂȚI FARMACEUTICE
- 7. DEȚINĂTORUL AUTORIZAȚIEI DE PUNERE PE PIAȚĂ
- 8. NUMĂRUL(ELE) AUTORIZAȚIEI DE PUNERE PE PIAȚĂ
- 9. DATA PRIMEI AUTORIZĂRI SAU A REÎNNOIRII AUTORIZAȚIEI
- 10. DATA REVIZUIRII TEXTULUI
1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI
Lazcluze 80 mg comprimate filmate
Lazcluze 240 mg comprimate filmate
2. COMPOZIȚIA CALITATIVĂ ȘI CANTITATIVĂ
Lazcluze 80 mg comprimate filmate
Fiecare comprimat filmat conține lazertinib 80 mg (sub formă de mesilat monohidrat).
Lazcluze 240 mg comprimate filmate
Fiecare comprimat filmat conține lazertinib 240 mg (sub formă de mesilat monohidrat).
Pentru lista tuturor excipienților, vezi pct. 6.1.
3. FORMA FARMACEUTICĂ
Comprimat filmat.
Lazcluze 80 mg comprimate filmate
Comprimat oval galben, de 14 mm, marcat cu „LZ” pe o față și cu „80” pe cealaltă față.
Lazcluze 240 mg comprimate filmate
Comprimat oval mov roșiatic, de 20 mm, marcat cu „LZ” pe o față și cu „240” pe cealaltă față.
4. DATE CLINICE
4.1 Indicații terapeutice
Lazcluze în asociere cu amivantamab este indicat în tratamentul de primă linie la pacienți adulți cu cancer pulmonar fără celule mici (NSCLC) în stadiu avansat, cu deleții la nivelul exonului 19 al EGFR sau mutații de substituție L858R la nivelul exonului 21 al EGFR.
4.2 Doze și mod de administrare
Tratamentul cu Lazcluze trebuie inițiat de un medic cu experiență în utilizarea medicamentelor antineoplazice.
Înainte de inițierea tratamentului cu Lazcluze, statusul pozitiv al mutației EGFR în probele de țesut tumoral sau plasmă trebuie stabilit utilizând o metodă de testare validată. În cazul în care nu se detectează nicio mutație într-o probă de plasmă, trebuie testat țesutul tumoral, dacă este disponibil într-o cantitate suficientă și are o calitate adecvată, din cauza potențialului de rezultate fals negative la utilizarea unui test bazat pe plasmă.
Doze
Doza recomandată de Lazcluze este de 240 mg o dată pe zi în asociere cu amivantamab.
Se recomandă administrarea de Lazcluze oricând înainte de amivantamab atunci când sunt administrate în aceeași zi. Pentru informațiile privind dozele recomandate de amivantamab, vezi pct. 4.2 din Rezumatul caracteristicilor produsului pentru amivantamab.
Evenimente tromboembolice venoase (TEV) asociate cu administrarea concomitentă cu amivantamab
La inițierea tratamentului, se recomandă administrarea profilactică de anticoagulante pacienților tratați cu Lazcluze în asociere cu amivantamab, pentru a preveni evenimentele tromboembolice venoase (TEV). Conform ghidurilor clinice, pacienților trebuie să li se admnistreze tratament profilactic fie cu un anticoagulant oral cu acțiune directă (AOAD), fie cu o heparină cu masă moleculară mică (HMMM). Utilizarea antagoniștilor vitaminei K nu este recomandată.
Reacții la nivelul pielii și unghiilor
Se recomandă terapia profilactică cu antibiotice cu administrare orală și topică pentru a reduce riscul de apariție și severitatea reacțiilor la nivelul pielii și unghiilor la pacienții cărora li se administrează Lazcluze în asociere cu amivantamab. Se recomandă, de asemenea, utilizarea unei creme hidratante necomedogene (de preferat pe bază de ceramide sau alte formule care asigură hidratarea îndelungată a pielii și nu conțin agenți de uscare) pe față și pe întreg corpul (cu excepția scalpului) și a unei soluții de clorhexidină pentru spălarea mâinilor și a picioarelor. Pacienții trebuie sfătuiți să reducă expunerea la soare pe durata tratamentului cu Lazcluze în asociere și timp de 2 luni după acesta. Pentru mai multe informații privind profilaxia reacțiilor la nivelul pielii și unghiilor, vezi pct. 4.4.
Durata tratamentului
Tratamentul trebuie continuat până la progresia bolii sau până la apariția toxicității inacceptabile.
Doza omisă
Dacă este omisă o doză planificată de Lazcluze, aceasta poate fi administrată în decurs de cel mult 12 ore. Dacă au trecut mai mult de 12 ore din momentul în care trebuia administrată doza, doza omisă nu trebuie administrată, iar următoarea doză trebuie administrată conform schemei obișnuite de administrare.
Modificări ale dozei
Recomandările privind reducerea dozelor din cauza reacțiilor adverse sunt prezentate în Tabelul 1.
Tabelul 1: Recomandări privind reducerea dozelor de Lazcluze din cauza reacțiilor adverse
| Reducerea dozei | Doza recomandată |
|---|---|
| Doza inițială | 240 mg o dată pe zi |
| Prima reducere a dozei | 160 mg o dată pe zi |
| A 2-a reducere a dozei | 80 mg o dată pe zi |
| A 3-a reducere a dozei | Se oprește tratamentul cu Lazcluze |
Modificările dozelor din cauza anumitor reacții adverse sunt prezentate în Tabelul 2.
Pentru informații privind modificările dozelor de amivantamab, vezi pct. 4.2 din Rezumatul caracteristicilor produsului pentru amivantamab.
Tabelul 2: Recomandări privind modificarea dozelor de Lazcluze și amivantamab din cauza reacțiilor adverse*
| Reacție adversă | Severitate | Modificarea dozei |
|---|---|---|
| Boală pulmonară interstițială (BPI)/pneumonită | Orice grad |
|
| Evenimente tromboembolice venoase (TEV) (vezi pct. 4.4) | Evenimente cu instabilitate clinică (de exemplu, insuficiență respiratorie sau disfuncție cardiacă) |
|
| Eveniment TEV recurent, în ciuda anticoagulării la nivel terapeutic |
| |
| Reacții la nivelul pielii și unghiilor (vezi pct. 4.4) | Grad 1 |
|
| Grad 2 |
| |
| Grad 3 |
Lazcluze cât și cu amivantamab. | |
| Grad 4 (inclusiv afecțiuni cutanate severe buloase, veziculare sau exfoliante, de exemplu, necroliză epidermică toxică) |
| |
| Hepatotoxicitate | Grad 3-4 |
|
| Parestezie | Grad 3-4 |
Lazcluze dacă recuperarea nu se obține în decurs de 4 săptămâni. |
| Diaree | Grad 3 |
|
| Grad 4 |
| |
| Stomatită | Grad 3-4 |
|
| Alte reacții adverse | Grad 3-4 |
|
*
Pentru informații privind dozele recomandate de amivantamab, vezi pct. 4.2 din Rezumatul caracteristicilor produsului pentru amivantamab.
Grupe speciale de pacienți
Vârstnici
Nu este necesară ajustarea dozei (vezi pct. 4.8, 5.1 și 5.2).
Insuficiența renală
Pe baza analizelor farmacocinetice (FC) populaționale, nu este necesară ajustarea dozei la pacienții cu insuficiență renală ușoară, moderată sau severă. Datele la pacienții cu insuficiență renală severă sunt limitate. Farmacocinetica lazertinibului la pacienții cu boală renală în stadiu terminal nu este cunoscută. Este necesară precauție la pacienții cu boală renală în stadiu terminal (vezi pct. 5.2).
Insuficiența hepatică
Nu este necesară ajustarea dozei la pacienții cu insuficiență hepatică ușoară sau moderată. Farmacocinetica lazertinibului la pacienții cu insuficiență hepatică severă nu este cunoscută. Este necesară precauție la pacienții cu insuficiență hepatică severă (vezi pct. 5.2).
Copii și adolescenți
Lazertinib nu prezintă utilizare relevantă la copii și adolescenți în tratamentul cancerului pulmonar fără celule mici.
Mod de administrare
Lazcluze este destinat administrării orale. Comprimatele trebuie înghițite întregi, cu sau fără alimente. Comprimatele nu trebuie zdrobite, divizate sau mestecate.
Dacă apar vărsături oricând după administrarea Lazcluze, următoarea doză trebuie luată în ziua următoare.
4.3 Contraindicații
Hipersensibilitate la substanța(ele) activă(e) sau la oricare dintre excipienții enumerați la pct. 6.1.
4.4 Atenționări și precauții speciale pentru utilizare
Boală pulmonară interstițială/pneumonită
La pacienții cărora li s-a administrat lazertinib și amivantamab au fost raportate cazuri de boală pulmonară interstițială (BPI) și reacții adverse similare BPI (de exemplu pneumonită), inclusiv evenimente letale (vezi pct. 4.8). Pacienții cu antecedente medicale de BPI, BPI indusă de medicamente, pneumonită de iradiere care a necesitat tratament cu steroizi, sau orice dovadă de BPI activă clinic au fost excluși din studiul clinic pivot.
Pacienții trebuie monitorizați pentru depistarea simptomelor care indică BPI/pneumonită (de exemplu, dispnee, tuse, febră). Dacă apar simptome, tratamentul cu Lazcluze trebuie întrerupt cât timp sunt investigate simptomele. Suspiciunea de BPI sau reacțiile adverse similare BPI trebuie evaluate și trebuie inițiat tratamentul adecvat, după caz. Tratamentul cu Lazcluze trebuie oprit permanent la pacienții cu BPI confirmată sau cu reacții adverse similare BPI (vezi pct. 4.2).
Evenimente tromboembolice venoase (TEV)
La pacienții cărora li s-a administrat Lazcluze în asociere cu amivantamab au fost raportate evenimente tromboembolice venoase (TEV), inclusiv tromboză venoasă profundă (TVP) și embolie pulmonară (EP), inclusiv evenimente cu evoluție letală (vezi pct. 4.8). Conform ghidurilor clinice, pacienților trebuie să li se administreze preventiv fie un anticoagulant oral cu acțiune directă (AOAD), fie o heparină cu masă moleculară mică (HMMM). Utilizarea antagoniștilor vitaminei K nu este recomandată.
Trebuie monitorizate semnele și simptomele de evenimente TEV. Pacienții cu evenimente TEV trebuie tratați cu anticoagulante, după cum este indicat clinic. Pentru evenimentele TEV asociate cu instabilitate clinică, tratamentul trebuie amânat până când pacientul este stabil clinic. După aceea, administrarea ambelor medicamente poate fi reluată, cu aceeași doză.
În cazul recurenței apărute în ciuda tratamentului anticoagulant corespunzător, tratamentul cu amivantamab trebuie oprit definitiv. Tratamentul poate continua cu Lazcluze, cu aceeași doză (vezi pct. 4.2).
Reacții la nivelul pielii și unghiilor
La pacienții tratați cu lazertinib în asociere cu amivantamab au apărut erupție cutanată tranzitorie (inclusiv dermatită acneiformă), prurit și xerodermie (vezi pct. 4.8). Pacienții trebuie sfătuiți să reducă expunerea la soare pe durata tratamentului cu Lazcluze în asociere și timp de 2 luni după acesta. Se recomandă purtarea unor haine de protecție și utilizarea unor produse de protecție cu spectru larg împotriva UVA/UVB.. Este recomandată profilaxia pentru prevenirea erupției cutanate tranzitorii. Aceasta include terapie profilactică, la inițierea tratamentului, cu un antibiotic cu administrare orală (de exemplu doxiciclină sau minociclină, 100 mg, de două ori pe zi) începând cu Ziua 1, în primele 12 săptămâni de tratament, iar după finalizarea terapiei cu antibiotic cu administrare orală, utilizarea topică la nivelul scalpului a unei loțiuni cu antibiotic (de exemplu clindamicină 1%) pentru următoarele 9 luni de tratament. Este recomandată utilizarea unei creme hidratante necomedogene (sunt preferate formulele pe bază de ceramide sau alte formule care asigură hidratarea îndelungată a pielii și nu conțin agenți de uscare) pe față și pe întregul corp (cu excepția scalpului) și a unei soluții cu clorhexidină pentru spălarea mâinilor și picioarelor, începând cu Ziua 1 și în continuare în timpul tratamentului.
Se recomandă ca la momentul administrării dozei inițiale să fie prescrise antibiotice cu utilizare topică și/sau orală și corticosteroizi topici, pentru a reduce la minimum orice întârziere în abordarea terapeutică a erupției cutanate tranzitorii, în cazul în care aceasta se dezvoltă în ciuda tratamentului profilactic. Dacă apar reacții la nivelul pielii sau unghiilor, trebuie administrată terapie de susținere, precum și corticosteroizi topici și antibiotice cu utilizare topică și/sau orală. De asemenea, pentru evenimente de gradul 3 sau evenimente de gradul 2 cu toleranță redusă, trebuie administrate antibiotice sistemice și steroizi cu utilizare orală și trebuie luat în considerare un consult dermatologic. Administrarea Lazcluze trebuie efectuată cu doză redusă, întreruptă sau oprită definitiv, în funcție de severitate (vezi pct. 4.2).
Tulburări oculare
La pacienții tratați cu lazertinib în asociere cu amivantamab au apărut tulburări oculare, inclusiv cheratită (vezi pct. 4.8). Pacienții care prezintă o agravare a simptomelor oculare trebuie trimiși imediat la oftalmolog și trebuie să întrerupă utilizarea lentilelor de contact până la evaluarea simptomelor.
Excipienți
Acest medicament conține sodiu mai puțin de 1 mmol (23 mg) per comprimat, adică practic „nu conține sodiu”.
4.5 Interacțiuni cu alte medicamente și alte forme de interacțiune
Inductorii puternici ai CYP3A4 pot determina scăderea concentrațiile plasmatice de lazertinib. Lazertinib poate crește concentrațiile plasmatice ale substraturilor CYP3A4 și BCRP.
Agenți care pot modifica concentrațiile plasmatice de lazertinib
Inductori ai CYP3A4
La subiecții sănătoși, administrarea concomitentă a mai multor doze de rifampicină (un inductor puternic al CYP3A4) a scăzut Cmax a lazertinibului cu 72% și ASC cu 83%. Administrarea de Lazcluze concomitent cu inductori puternici ai CYP3A4 (de exemplu carbamazepină, fenitoină, rifampicină, sunătoare) trebuie evitată. De asemenea, administrarea concomitentă de Lazcluze cu inductori moderați ai CYP3A4 (de exemplu bosentan, efavirenz, modafinil) poate scădea concentrațiile plasmatice de lazertinib, prin urmare inductorii moderați ai CYP3A4 trebuie utilizați cu precauție.
Inhibitori ai CYP3A4
La subiecții sănătoși, administrarea concomitentă a mai multor doze de itraconazol (un inhibitor puternic al CYP3A4) a crescut Cmax de 1,19 ori și ASC de 1,46 ori. Nu este necesară ajustarea dozei inițiale atunci când Lazcluze este administrat concomitent cu inhibitori ai CYP3A4.
Agenți care reduc producerea de acid gastric
Nu au fost observate diferențe relevante clinic în farmacocinetica lazertinibului atunci când a fost administrat concomitent cu agenți care reduc producerea de acid gastric (inhibitori ai pompei de protoni și antagoniști ai receptorului H2). Nu sunt necesare ajustări ale dozei atunci când Lazcluze este utilizat concomitent cu agenți care reduc producerea de acid gastric.
Agenți ale căror concentrații plasmatice pot fi modificate de Lazcluze
Substraturi ale CYP3A4
Administrarea concomitentă a mai multor doze de Lazcluze 160 mg o dată pe zi a crescut Cmax a midazolamului (un substrat al CYP3A4) de 1,39 ori și ASC de 1,47 ori. Medicamentele cu indice terapeutic îngust care sunt substraturi ale CYP3A4 (de exemplu, ciclosporină, everolimus, pimozidă, chinidină, sirolimus, tacrolimus) trebuie utilizate cu precauție, deoarece lazertinib poate crește concentrațiile plasmatice ale acestor medicamente.
Substraturi ale BCRP
Administrarea concomitentă a mai multor doze de Lazcluze 160 mg o dată pe zi a crescut Cmax a rosuvastatinei (un substrat al BCRP) de 2,24 ori și ASC de 2,02 ori. Medicamentele cu indice terapeutic îngust care sunt substraturi ale BCRP (de exemplu, sunitinib) trebuie utilizate cu precauție, deoarece lazertinib poate crește concentrațiile plasmatice ale acestor medicamente.
Substraturi ale CYP1A2
Inducerea CYP1A2 nu poate fi exclusă. Prin urmare, se recomandă prudență atunci când se administrează concomitent cu substraturi ale CYP1A2 (de exemplu, tizanidină).
4.6 Fertilitatea, sarcina și alăptarea
Femei aflate la vârsta fertilă/Contracepția la bărbați și femei
Femeile aflate la vârsta fertilă trebuie să utilizeze măsuri contraceptive eficace în timpul și timp de până la 3 săptămâni după tratament.
Pacienții de sex masculin trebuie sfătuiți să utilizeze măsuri contraceptive eficace (de exemplu, prezervativ) și să nu doneze sau să păstreze spermă în timpul tratamentului și timp de 3 săptămâni după ultima doză de lazertinib.
Sarcina
Datele provenind din utilizarea lazertinibului la femeile gravide sunt inexistente. Studiile la animale au evidențiat efecte toxice asupra funcției de reproducere (supraviețuire embrio-fetală redusă și greutate corporală mai scăzută a fetusului) (vezi pct. 5.3). Pe baza mecanismului său de acțiune și a datelor provenind de la animale, lazertinib poate avea efecte nocive asupra fătului atunci când este administrat femeilor gravide. Lazertinibul nu trebuie utilizat în cursul sarcinii, cu excepția cazului în care se consideră că beneficiile tratamentului pentru femeie depășesc riscurile potențiale pentru făt. Dacă rămâne gravidă în timp ce ia acest medicament, pacienta trebuie informată cu privire la riscul potențial pentru făt.
Alăptarea
Nu se cunoaște dacă lazertinib sau metaboliții acestuia se excretă în laptele uman sau dacă afectează producerea de lapte. Deoarece nu se poate exclude un risc pentru copilul alăptat, pacientele trebuie sfătuite să nu alăpteze în timpul tratamentului și timp de 3 săptămâni după ultima doză de lazertinib.
Fertilitatea
Datele privind efectul Lazcluze asupra fertilității la om sunt inexistente. Studiile la animale au arătat că lazertinib are efecte asupra organelor de reproducere la femele (număr scăzut de cicluri estrale și de corpi luteali) și la masculi (modificări degenerative ale testiculelor) și poate afecta fertilitatea la femei și bărbați (vezi pct. 5.3).
4.7 Efecte asupra capacității de a conduce vehicule și de a folosi utilaje
Lazcluze are influență minoră asupra capacității de a conduce vehicule sau de a folosi utilaje. Dacă pacienții prezintă simptome legate de tratament (precum fatigabilitate), care le afectează capacitatea de concentrare și reacție, se recomandă ca aceștia să nu conducă vehicule sau să folosească utilaje până când efectul dispare.
4.8 Reacții adverse
Rezumatul profilului de siguranță
Cele mai frecvente reacții adverse de toate gradele au fost erupție cutanată tranzitorie (89%), toxicitate la nivelul unghiilor (71%), reacție adversă legată de perfuzie (amivantamab) (63%), hipoalbuminemie (amivantamab) (48%), hepatotoxicitate (47%), edem (amivantamab) (47%), stomatită (43%), tromboembolism venos (36%), parestezie (34%), fatigabilitate (32%), constipație (29%), diaree (29%), xerodermie (26%), apetit alimentar scăzut (24%), prurit (24%), hipocalcemie (21%), alte tulburări oculare (21%) și greață (21%).
Cele mai frecvente reacții adverse grave au inclus tromboembolism venos (11%), pneumonie (4,0%), erupții cutanate (3,1%), boală pulmonară interstițială/pneumonită (2,9%), COVID-19 (2,4%), hepatotoxicitate (2,4%), efuziune pleurală (2,1%), reacție adversă legată de perfuzie (amivantamab) (2,1%), insuficiență respiratorie (1,4%), fatigabilitate (1,2%), edem (amivantamab) (1,2%), hipoalbuminemie (amivantamab) (1,2%) și hiponatremie (1,2%).
La pacienții cărora li s-a administrat Lazcluze în asociere cu amivantamab, cele mai frecvente reacții adverse care au dus la oprirea administrării oricăreia dintre substanțele din cadrul tratamentului au fost erupțiile cutanate (6%), reacție adversă legată de perfuzie (amivantamab) (4,5%), toxicitate la nivelul unghiilor (3,6%), boală pulmonară interstițială/pneumonită (2,9%), tromboembolism venos (2,9%), pneumonie (1,9%) și edem (amivantamab) (1,7%).
Lista tabelară a reacțiilor adverse
Tabelul 3 prezintă rezumatul reacțiilor adverse apărute la pacienții tratați cu lazertinib în asociere cu amivantamab.
Datele reflectă expunerea la lazertinib a 421 de pacienți cărora li s-a administrat lazertinib în asociere cu amivantamab în studiul MARIPOSA. Mediana duratei de expunere la lazertinib a fost de 18,5 luni (interval: 0,2 până la 31,4 luni).
Reacțiile adverse observate în timpul studiilor clinice sunt enumerate mai jos în funcție de frecvență. Frecvențele sunt definite după cum urmează: foarte frecvente (≥1/10), frecvente (≥1/100 și < 1/10), mai puțin frecvente (≥1/1 000 și < 1/100), rare (≥1/10 000 și < 1/1 000), foarte rare (< 1/10 000) și cu frecvență necunoscută (care nu poate fi estimată din datele disponibile). În cadrul fiecărei grupe de frecvență, reacțiile adverse sunt prezentate în ordinea descrescătoare a gravității.
Tabelul 3: Reacții adverse la pacienții tratați cu lazertinib în asociere cu amivantamab
| Clasificare pe aparate, sisteme și organe Reacție adversă | Categoria de frecvență | Orice grad (%) | Grad 3-4 (%) |
|---|---|---|---|
| Tulburări metabolice și de nutriție | |||
| Hipoalbuminemiea,b | Foarte frecvente | 48 | 5 |
| Apetit alimentar scăzut | 24 | 1,0 | |
| Hipocalcemie | 21 | 2,1 | |
| Hipopotasemie | 14 | 3,1 | |
| Hipomagneziemie | Frecvente | 5 | 0 |
| Tulburări ale sistemului nervos | |||
| Paresteziea | Foarte frecvente | 34 | 1,7 |
| Amețealăa | 13 | 0 | |
| Tulburări oculare | |||
| Alte tulburări ocularea | Foarte frecvente | 21 | 0,5 |
| Tulburări de vederea | Frecvente | 4,5 | 0 |
| Cheratită | 2,6 | 0,5 | |
| Creștere a genelora | 1,9 | 0 | |
| Tulburări vasculare | |||
| Tromboembolism venosa | Foarte frecvente | 37 | 11 |
| Tulburări respiratorii, toracice și mediastinale | |||
| Boală pulmonară interstițială/pneumonităa | Frecvente | 3,1 | 1,2 |
| Tulburări gastro-intestinale | |||
| Stomatităa | Foarte frecvente | 43 | 2,4 |
| Diaree | 29 | 2,1 | |
| Constipație | 29 | 0 | |
| Greață | 21 | 1,2 | |
| Vărsături | 12 | 0,5 | |
| Dureri abdominalea | 11 | 0 | |
| Hemoroizi | Frecvente | 10 | 0,2 |
| Tulburări hepatobiliare | |||
| Hepatotoxicitatea | Foarte frecvente | 47 | 9 |
| Afecțiuni cutanate și ale țesutului subcutanat | |||
| Erupție cutanată tranzitoriea | Foarte frecvente | 89 | 27 |
| Toxicitate la nivelul unghiilora | 71 | 11 | |
| Xerodermiea | 26 | 1,0 | |
| Prurit | 24 | 0,5 | |
| Sindrom de eritrodisestezie palmo-plantară | Frecvente | 6 | 0,2 |
| Urticarie | 1,2 | 0 | |
| Tulburări musculo-scheletice și ale țesutului conjunctiv | |||
| Spasme musculare | Foarte frecvente | 17 | 0,5 |
| Mialgie | 13 | 0,7 | |
| Tulburări generale și la nivelul locului de administrare | |||
| Edema, b | Foarte frecvente | 47 | 2,9 |
| Fatigabilitatea | 32 | 3,8 | |
| Pirexie | 12 | 0 | |
| Leziuni, intoxicații și complicații legate de proceduri | |||
| Reacție adversă legată de perfuzieb | Foarte frecvente | 63 | 6 |
a termeni grupați b aplicabil numai pentru amivantamab.
Descrierea anumitor reacții adverse
Tromboembolism venos
La 37% dintre pacienții cărora li s-a administrat lazertinib în asociere cu amivantamab au fost raportate evenimente tromboembolice venoase (TEV), inclusiv tromboză venoasă profundă (14,5%) și embolie pulmonară (EP) (17,3%). Majoritatea evenimentelor au fost de gradul 1 sau 2, evenimentele de gradul 3-4 survenind la 11%, iar decesele la 0,5% dintre pacienții cărora li s-a administrat lazertinib în asociere cu amivantamab. Pentru informații privind profilaxia cu anticoagulante și abordarea terapeutică a evenimentelor TEV, vezi pct. 4.2 și 4.4.
La pacienții tratați cu lazertinib în asociere cu amivantamab, mediana intervalului de timp până la primul debut al evenimentului TEV a fost de 84 zile. Evenimentele TEV au dus la oprirea administrării oricăreia dintre substanțele din cadrul tratamentului la 2,9% dintre pacienți.
Boală pulmonară interstițială (BPI)/pneumonită
Au fost raportate boala pulmonară interstițială sau reacții adverse similare cu BPI (de exemplu, pneumonită) în legătură cu utilizarea de lazertinib în asociere cu amivantamab, precum și cu alți inhibitori EGFR. La 3,1% dintre pacienții tratați cu lazertinib în asociere cu amivantamab a fost raportată BPI sau pneumonita, inclusiv 0,2% cazuri cu evoluție letală. Pacienții cu antecedente de BPI, BPI indusă de medicamente, pneumonită de iradiere care a necesitat tratament cu steroizi sau cu orice dovezi de BPI clinic activă au fost excluși din studiul clinic (vezi pct. 4.4).
Reacții la nivelul pielii și unghiilor
Au apărut erupție cutanată tranzitorie (inclusiv dermatită acneiformă), prurit și xerodermie. La 89% dintre pacienții tratați cu lazertinib în asociere cu amivantamab a apărut erupția cutanată tranzitorie. Majoritatea cazurilor au fost de gradul 1 sau 2, evenimentele de gradul 3 survenind la 27% dintre pacienți. La 6% dintre pacienți a apărut erupție cutanată care a dus la oprirea administrării oricăreia dintre substanțele din cadrul tratamentului. Erupția cutanată tranzitorie a apărut de regulă în primele 4 săptămâni de tratament, cu o mediană a intervalului de timp până la debut de 14 zile. La pacienții tratați cu lazertinib în asociere cu amivantamab a apărut toxicitate la nivelul unghiilor. Majoritatea evenimentelor au fost de gradul 1 sau 2, evenimentele de toxicitate la nivelul unghiilor de gradul 3 survenind la 11% dintre pacienți (vezi pct. 4.4).
A fost realizat un studiu de fază 2 la pacienții tratați cu Lazcluze în asociere cu amivantamab pentru a evalua utilizarea terapiei profilactice cu un antibiotic cu utilizare orală, un antibiotic administrat topic la nivelul scalpului, o cremă hidratantă pe față și pe întreg corpul (cu excepția scalpului) și un antiseptic pe mâini și picioare (vezi pct. 4.2 și 4.4). S-a demonstrat o reducere a incidenței evenimentelor adverse dermatologice de grad ≥ 2 în primele 12 săptămâni de tratament, comparativ cu tratamentul dermatologic standard utilizat în practica clinică (38,6% față de 76,5%, p<0,0001). În plus, s-a observat o reducere a evenimentelor adverse de grad ≥ 2 care au afectat scalpul în primele 12 săptămâni de tratament (8,6% față de 29,4%), împreună cu o incidență mai mică a reducerilor de doză (7,1% față de 19,1%), a întreruperilor administrării (15,7% față de 33,8%) și a opririi tratamentului (1,4% față de 4,4%) din cauza evenimentelor adverse dermatologice.
Tulburări oculare
La pacienții tratați cu lazertinib în asociere cu amivantamab au apărut tulburări oculare, inclusiv cheratită (2,6%). Alte reacții adverse raportate au inclus creștere a genelor, tulburări de vedere și alte tulburări oculare. Majoritatea evenimentelor au fost de gradul 1-2 (vezi pct. 4.4).
Hepatotoxicitate
Reacțiile legate de hepatotoxicitate au apărut la 47% dintre pacienții tratați cu lazertinib în asociere cu amivantamab. Majoritatea evenimentelor au fost de gradul 1-2, hepatotoxicitatea de gradul 3-4 apărând la 9% dintre pacienți. Majoritatea evenimentelor au fost legate de creșterea valorilor serice ale transaminazelor (36% valori serice crescute ale alanin aminotransferazei și 29% valori serice crescute ale aspartat aminotransferazei). Majoritatea pacienților cu creșteri ale valorilor serice ale transaminazelor au putut continua tratamentul de studiu, fără modificarea acestuia, în timp ce la un număr mic s-a luat decizia de a întrerupe administrarea sau de a reduce doza. În studiile clinice cu lazertinib în asociere cu amivantamab, nu au existat cazuri de insuficiență hepatică sau cazuri de hepatotoxicitate letale.
Au fost raportate cazuri izolate de creștere a valorilor fosfatazei alcaline și valori persistent crescute ale bilirubinei în cadrul monoterapiei cu lazertinib.
Parestezie
Parestezia a apărut la 34% dintre pacienții tratați cu lazertinib în asociere cu amivantamab. Majoritatea evenimentelor au fost de gradul 1-2, parestezia de gradul 3 apărând la 1,7% dintre pacienți. La majoritatea pacienților cu parestezie s-a luat decizia de a întrerupe administrarea sau de a reduce doza.
Stomatită
Stomatita a apărut la 43% dintre pacienții tratați cu lazertinib în asociere cu amivantamab. Majoritatea evenimentelor au fost de gradul 1-2, stomatita de gradul 3 apărând la 2,4% dintre pacienți.
Diaree
Diareea a apărut la 29% dintre pacienții tratați cu lazertinib în asociere cu amivantamab. Majoritatea evenimentelor au fost de gradul 1-2, diareea de gradul 3 apărând la 2,1% dintre pacienți.
Grupe speciale de pacienți
Vârstnici
Datele clinice privind utilizarea de lazertinib la pacienți cu vârsta de 75 de ani sau peste sunt limitate (vezi pct. 5.1). Pacienții vârstnici (≥65 ani) au raportat mai multe evenimente adverse de gradul 3 sau mai mare, comparativ cu pacienții cu vârsta < 65 ani (81% față de 70%). În timp ce ratele de întrerupere a administrării medicamentelor și de reducere a dozei au fost similare, rata evenimentelor adverse care au dus la oprirea administrării oricăreia dintre substanțele din cadrul tratamentului a fost mai mare la pacienții cu vârsta ≥65 ani, comparativ cu pacienții cu vârsta < 65 ani (47% față de 25%).
Raportarea reacțiilor adverse suspectate
Raportarea reacțiilor adverse suspectate după autorizarea medicamentului este importantă. Acest lucru permite monitorizarea continuă a raportului beneficiu/risc al medicamentului. Profesioniștii din domeniul sănătății sunt rugați să raporteze orice reacție adversă suspectată prin intermediul sistemului național de raportare, astfel cum este menționat în Anexa V.
4.9 Supradozaj
Nu există antidot cunoscut în caz de supradozaj cu Lazcluze. În caz de supradozaj, se oprește tratamentul cu Lazcluze și se iau măsurile generale de susținere. Pacienții trebuie monitorizați atent pentru depistarea semnelor sau simptomelor reacțiilor adverse.
5. PROPRIETĂȚI FARMACOLOGICE
5.1 Proprietăți farmacodinamice
Grupa farmacoterapeutică: medicamente antineoplazice, inhibitori de protein kinază, codul ATC: L01EB09.
Mecanism de acțiune
Lazertinib este un inhibitor ireversibil al tirozin kinazei (TKI) EGFR. Acesta inhibă selectiv atât mutațiile de activare primară din domeniul EGFR (deleții la nivelul exonului 19 și mutații de substituție L858R la nivelul exonului 21), cât și mutația de rezistență T790M din domeniul EGFR, având în același timp o activitate mai redusă împotriva EGFR de tip sălbatic.
Efecte farmacodinamice
Pe baza analizelor expunere-răspuns pentru siguranță, parestezia și stomatita au părut să prezinte o tendință de creștere a incidenței, odată cu creșterea expunerii la lazertinib.
Electrofiziologie cardiacă
Potențialul de prelungire a intervalului QTc al lazertinibului a fost evaluat prin analiza expunererăspuns (E-R), efectuată cu date clinice de la 243 de pacienți cu NSCLC, cărora li s-a administrat lazertinib în doză de 20, 40, 80, 120, 160, 240 sau 320 mg o dată pe zi într-un studiu de fază 1/II. Analiza E-R nu a relevat nicio legătură relevantă clinic între concentrația plasmatică de lazertinib și modificarea intervalului QTc.
Eficacitate și siguranță clinică
MARIPOSA este un studiu multicentric de fază 3, randomizat, în regim deschis, controlat activ, de evaluare a eficacității și siguranței Lazcluze în asociere cu amivantamab, comparativ cu osimertinib în monoterapie în tratamentul de primă linie la pacienții cu NSCLC cu mutații EGFR, avansat local sau metastazat, care nu răspunde la terapia curativă. A fost necesar ca probele pacienților să aibă una dintre cele două mutații comune din domeniul EGFR (deleție la nivelul exonului 19 sau mutație de substituție L858R la nivelul exonului 21), identificată prin testare la nivel local. Probele de țesut tumoral (94%) și/sau de plasmă (6%) prelevate de la toți pacienții au fost testate la nivel local pentru a determina statutul deleției la nivelul exonului 19 al EGFR și/sau al mutației de substituție L858R la nivelul exonului 21, utilizând reacția de polimerizare în lanț (polymerase chain reaction, PCR) la 65% dintre pacienți și secvențierea de generație următoare (NGS) la 35% dintre pacienți.
În total, 1074 de pacienți au fost randomizați (în raport de 2:2:1) pentru a li se administra Lazcluze în asociere cu amivantamab, osimertinib în monoterapie sau Lazcluze în monoterapie, până la progresia bolii sau până la toxicitate inacceptabilă. Lazcluze a fost administrat oral în doză de 240 mg o dată pe zi. Amivantamab a fost administrat intravenos în doză de 1050 mg (pentru pacienții cu greutate < 80 kg) sau 1400 mg (pentru pacienții cu greutate ≥80 kg) o dată pe săptămână timp de 4 săptămâni și ulterior, începând cu săptămâna 5, la interval de 2 săptămâni. Osimertinib a fost administrat oral în doză de 80 mg o dată pe zi. Randomizarea a fost stratificată în funcție de tipul mutației din domeniul EGFR (deleție la nivelul exonului 19 sau mutație de substituție L858R la nivelul exonului 21), rasă (asiatică sau non-asiatică) și antecedente de metastaze cerebrale (da sau nu).
Caracteristicile demografice și ale bolii la momentul inițial au fost echilibrate între grupurile de tratament. Mediana vârstei a fost de 63 de ani (interval: 25-88), 45% dintre pacienți având vârsta ≥65 de ani și 11% vârsta ≥75 de ani; 62% au fost femei; 59% au fost asiatici și 38% caucazieni. Statusul de performanță al Grupului Estic pentru Cooperare în Oncologie (Eastern Cooperative Oncology Group, ECOG) a fost 0 (34%) sau 1 (66%); 69% nu fumaseră niciodată; 41% aveau metastaze cerebrale anterioare, iar 90% aveau cancer în stadiul IV la momentul diagnosticului inițial. În ceea ce privește statusul mutațiilor în domeniul EGFR, 60% erau deleții la nivelul exonului 19 și 40% erau mutații de substituție L858R la nivelul exonului 21.
Lazcluze în asociere cu amivantamab a demonstrat o îmbunătățire semnificativă statistic a supraviețuirii fără progresia bolii (SFP), pe baza evaluării BICR.
Analiza finală privind SG a demonstrat o îmbunătățire semnificativă din punct de vedere statistic a SG în cazul tratamentului cu Lazcluze în asociere cu amivantamab în comparație cu tratamentul cu osimertinib (vezi Tabelul 4 și Figura 2).
Tabelul 4, Figura 1 și Figura 2 rezumă rezultatele privind eficacitatea pentru Lazcluze în asociere cu amivantamab.
Tabelul 4: Rezultate privind eficacitatea în studiul MARIPOSA
| Lazcluze + amivantamab (N=429) | Osimertinib (N=429) | |
| Supraviețuire fără progresia bolii (SFP)a | ||
|---|---|---|
| Număr de evenimente | 192 (45%) | 252 (59%) |
| Mediană, luni (IÎ 95%) | 23,7 (19,1, 27,7) | 16,6 (14,8, 18,5) |
| RR (IÎ 95%); valoarea p | 0,70 (0,58, 0,85); p=0,0002 | |
| Supraviețuire globală (SG) | ||
| Număr de evenimente | 173 (40%) | 217 (51%) |
| Mediana, luni (IÎ 95%) | NE (42,9, NE) | 36,7 (33,4, 41,0) |
| RR (IÎ 95%); valoarea p | 0,75 (0,61, 0,92); p=0.0048 | |
| Rata răspunsului obiectiv (RRO)a, b | ||
| % RRO (IÎ 95%) | 80% (76%, 84%) | 77% (72%, 81%) |
| Durata răspunsului (DR)a, b | ||
| Mediană, luni (IÎ 95%) | 25,8 (20,3, 33,9) | 18,1 (14,8, 20,1) |
BICR = analiză centrală independentă în regim orb; IÎ = interval de încredere; NE = nu se poate estima.
Rezultatele privind SFP se referă la data centralizării datelor 11 august 2023, cu o perioadă mediană de urmărire de 22,0 luni. Rezultatele privind RRO și DR se referă la data centralizării datelor 13 mai 2024, cu o perioadă mediană de urmărire de 31,3 luni. Rezultatele privind SG se referă la data centralizării datelor 4 decembrie 2024, cu o perioadă mediană de urmărire de 37,8 luni. a
BICR pe baza criteriilor RECIST v1.1. b
Pe baza respondenților confirmați.
Figura 1: Curba Kaplan-Meier a SFP la pacienții cu NSCLC netratați anterior, pe baza evaluării BICR

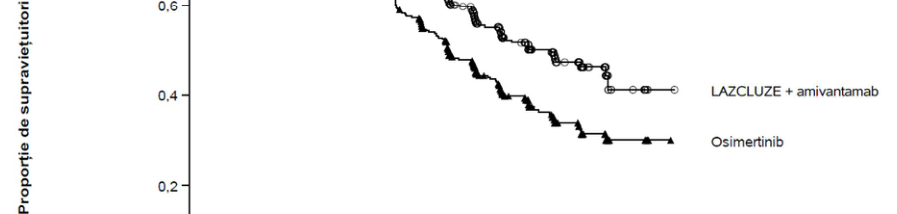

Figura 2: Curba Kaplan-Meier a SG la pacienții cu NSCLC netratați anterior



În studiul MARIPOSA, RRO intracranian și DR pe baza evaluării BICR au fost criterii finale de evaluare pre-specificate. În subsetul de pacienți cu leziuni intracraniene la momentul inițial, administrarea de Lazcluze în asociere cu amivantamab a demonstrat un RRO intracranian similar cu cel al substanței de control. Conform protocolului, toți pacienții din studiul MARIPOSA au fost supuși unei serii de examinări RMN, pentru a evalua răspunsul intracranian și durata. Rezultatele sunt prezentate pe scurt în Tabelul 5.
Tabelul 5: RRO intracranian și DR pe baza evaluării BICR la subiecții cu leziuni intracraniene la momentul inițial
| Lazcluze + amivantamab (N=180) | Osimertinib (N=186) | |
| Evaluarea răspunsului tumoral intracranian | ||
|---|---|---|
| RRO intracranian (RC+RP), % (IÎ 95%) | 78% (71%, 84%) | 77% (71%, 83%) |
| Răspuns complet | 64% | 59% |
| DR intracranian | ||
| Mediană, luni (IÎ 95%) | 35,0 (20,4, NE) | 25,1 (22,1, 31,2) |
IÎ = interval de încredere; NE = nu se poate estima
Rezultatele privind RRO intracranian și DR se referă la data centralizării datelor 4 decembrie 2024, cu o perioadă mediană de urmărire de 37,8 luni.
Copii și adolescenți
Agenția Europeană pentru Medicamente a acordat o derogare de la obligația de depunere a rezultatelor studiilor efectuate cu Lazcluze la toate subgrupurile de copii și adolescenți în cancerul pulmonar fără celule mici.
5.2 Proprietăți farmacocinetice
În urma administrării orale de doze unice și repetate o dată pe zi, concentrația plasmatică maximă a lazertinibului (Cmax) și aria de sub curba concentrației plasmatice în funcție de timp (ASC) au crescut aproximativ proporțional cu doza, în intervalul de doze de la 20 până la 320 mg.
Expunerea plasmatică la starea de echilibru a fost atinsă până în ziua 15 de administrare o dată pe zi și s-a observat o acumulare de aproximativ 2 ori la starea de echilibru în cazul utilizării dozei de 240 mg o dată pe zi.
Expunerea plasmatică la lazertinib a fost comparabilă atunci când lazertinib a fost administrat fie în asociere cu amivantamab, fie în monoterapie.
Absorbție
Mediana intervalului de timp până la atingerea Cmax în cazul utilizării unei dozei unice și a stării de echilibru în cazul utilizării de doze repetate a fost comparabilă și a variat între 2 și 4 ore.
În urma administrării de lazertinib 240 mg în timpul unei mese bogate în grăsimi (800-1000 kcal, conținut de grăsimi de aproximativ 50%), Cmax și ASC ale lazertinibului au fost comparabile cu cele observate în condiții de repaus alimentar, sugerând că lazertinibul poate fi administrat cu sau fără alimente.
Distribuție
Lazertinibul a fost distribuit extensiv, cu o valoare medie (CV%) a volumului aparent de distribuție de 4264 (43,2%) l în cazul utilizării dozei de 240 mg. Valoarea medie (CV%) a legării de proteinele plasmatice pentru lazertinib a fost de aproximativ 99,2% (0,13%) la om. Lazertinib a prezentat legare covalentă de proteinele din sângele și plasma umană după administrarea orală și în timpul incubărilor in vitro.
Metabolizare
Lazertinib este metabolizat în principal prin conjugarea glutationului, fie enzimatic prin glutation S-transferază (GST), fie non-enzimatic, precum și prin intermediul CYP3A4. Cei mai abundenți metaboliți sunt cataboliții glutationici și sunt considerați inactivi clinic. Expunerea plasmatică la lazertinib a fost afectată de metabolizarea mediată de GSTM1, ducând la o expunere mai redusă (o diferență mai mică decât dublu) la pacienții cu GSTM1 non-nul. Nu este necesară ajustarea dozei în funcție de statusul GSTM1.
Eliminare
Valoarea medie (CV%) a clearance-ului aparent și timpul de înjumătățire terminal ale lazertinibului administrat în doza de 240 mg au fost de 44,5 (29,5%) l/oră și, respectiv, 64,7 (32,8%) ore.
Excreție
După administrarea orală a unei doze unice de lazertinib marcat radioactiv, aproximativ 86% din doză s-a regăsit în materiile fecale (< 5% nemodificat) și 4% în urină (< 0,5% nemodificat).
Administrarea concomitentă cu substraturi ale OCT1 și UGT1A1
Administrarea concomitentă a mai multor doze de Lazcluze nu a crescut Cmax și ASC ale metformin (un substrat al OCT1). Lazcluze nu inhibă OCT1.
Pe baza studiilor in vitro, Lazcluze poate inhiba UGT1A1. Cu toate acestea, pe baza lipsei efectului asupra valorilor bilirubinemiei indirecte în studiul clinic, nu se așteaptă nicio interacțiune relevantă clinic cu substraturile UGT1A1.
Grupe speciale de pacienți
Vârstnici
Pe baza analizei farmacocineticii populaționale, nu au fost observate diferențe relevante clinic în funcție de vârstă în farmacocinetica lazertinbului.
Insuficiența renală
Pe baza analizei farmaconetice populaționale, nu este necesară ajustarea dozei la pacienții cu insuficiență renală ușoară, moderată sau severă, cu rata de filtrare glomerulară estimată (RFGe) de 15 până la 89 ml/minut. Datele referitoare la pacienții cu insuficiență renală severă (RFGe de 15 până la 29 ml/minut) sunt limitate (n=3), dar nu există dovezi care să sugereze că este necesară ajustarea dozei la acești pacienți. Nu sunt disponibile date referitoare la pacienții cu boală renală în stadiu terminal (RFGe < 15 ml/minut).
Insuficiența hepatică
Pe baza concluziilor studiului privind farmacologia clinică, insuficiența hepatică moderată (Child-Pugh Clasa B) nu a avut niciun efect relevant clinic asupra farmacocineticii dozei unice de lazertinib. Pe baza analizei farmacocinetice populaționale, nu este necesară ajustarea dozei la pacienții cu insuficiență hepatică ușoară (bilirubinemie totală ≤LSN și AST > LSN sau LSN < bilirubinemie totală ≤1,5 × LSN și orice valoare serică a AST) sau moderată (1,5 × LSN < bilirubinemie totală ≤3 × LSN și orice valoare serică a AST). Nu sunt disponibile datele privind pacienții cu insuficiență hepatică severă (bilirubinemie totală > 3 × LSN și orice valoare serică a AST).
Copii și adolescenți
Farmacocinetica lazertinibului la pacienții copii și adolescenți nu a fost investigată.
Alte grupe de pacienți
Nu s-au observat diferențe semnificative din punct de vedere clinic în ceea ce privește farmacocinetica lazertinibului în funcție de sex, greutate corporală, rasă, origine etnică, evaluări de laborator la momentul inițial (clearance-ul creatininei, albumină, alanin aminotransferază, fosfatază alcalină, aspartat aminotransferază), statusul de performanță ECOG, tipul de mutație în domeniul EGFR, stadiul cancerului la momentul diagnosticului inițial, terapiile anterioare, metastazele cerebrale și istoricul de fumat.
5.3 Date preclinice de siguranță
Principalele rezultate observate în studiile privind toxicitatea după administrarea de doze repetate de lazertinib la șobolan și câine au inclus atrofie epitelială ușoară până la eroziuni degenerative, inflamație și necroză care afectează ochii (atrofie corneană), pielea (blană subțire și aspră, degenerare a foliculilor de păr, alopecie, ulcerație), ficatul (valori serice crescute ale enzimelor hepatice, hipertrofie a celulelor Kupffer și necroză hepatocelulară), plămânii (infiltrat de macrofage la nivel alveolar, inflamație pulmonară și hiperplazie a celulelor alveolare de tip II), rinichii (dilatare tubulară, necroză papilară, creștere a valorii azotului ureic, creștere a creatininemiei (numai la femele), creștere a valorilor fosforului anorganic și potasiului), GI (atrofie a epiteliului esofagian, estompare/fuziune a vilozităților în duoden și jejun, materii fecale lichide), sistemul reproducător (degenerare tubulară a testiculelor, hipospermie, scădere a numărului ciclurilor estrale și a numărului corpilor luteali, atrofie a uterului și a vaginului). Aceste rezultate au fost observate la animale la expuneri de 0,9-3,4 ori mai mari decât expunerile estimate la pacienții cărora li s-a administrat doza recomandată (240 mg) și au fost complet sau parțial rezolvate în etapele de recuperare. Inima a fost considerată un organ țintă numai la câini și afectarea a apărut la valori de expunere de 7 ori mai mari decât de expunerea așteptată la doza recomandată la om.
Carcinogenicitate și mutagenicitate
Nu s-au observat dovezi de genotoxicitate pentru lazertinib în testele de mutagenicitate bacteriană in vitro, de aberație cromozomială in vitro și la testele micronucleilor in vivo la șobolan. Nu au fost efectuate studii de lungă durată la animale pentru a evalua potențialul carcinogen al lazertinibului.
Toxicitate asupra funcției de reproducere
Pe baza studiilor la animale, fertilitatea masculină și feminină poate fi afectată de tratamentul cu lazertinib. Au fost prezente modificări degenerative la nivelul testiculelor șobolanilor și câinilor, care au dus la reducerea spermei luminale la câine în urma expunerii la lazertinib timp de 1 lună, la valori de expunere relevante din punct de vedere clinic. A fost observat număr scăzut de corpi luteali în ovarele femelelor de șobolan expuse la lazertinib timp de ≥1 lună, la valori de expunere relevante clinic. Într-un studiu privind fertilitatea și dezvoltarea embrionară timpurie la masculi și femele de șobolan, lazertinib a indus o scădere a numărului de cicluri estrale, o creștere a pierderilor postimplantare și scădere a numărului de pui vii, la valori de doză sau sub valorile de doză care au determinat expuneri similare cu expunerea clinică la om în cazul utilizării dozei recomandate de 240 mg.
Toxicitatea pentru dezvoltare a fost observată în studiile de dezvoltare embrio-fetală la șobolan și iepure. La șobolan, s-au observat scăderi ale greutății corporale fetale în asociere cu toxicitatea maternă, la o expunere maternă de aproximativ 4 ori mai mare decât expunerea clinică la om în cazul utilizării dozei de 240 mg. La iepure, s-a observat o incidență crescută a fuziunii oaselor craniene la fetus (arcada zigomatică fuzionată cu procesul maxilar) la expuneri materne cu mult sub expunerea clinică la om în cazul utilizării dozei de 240 mg.
6. PROPRIETĂȚI FARMACEUTICE
6.1 Lista excipienților
Nucleul comprimatului
Siliciu coloidal hidrofob
Croscarmeloză sodică (E468)
Celuloză microcristalină (E460 (i))
Manitol (E421)
Stearat de magneziu (E572)
Film
Lazcluze 80 mg comprimate filmate
Copolimer grefat de alcool polivinilic și macrogol (E1209)
Alcool polivinilic (E1203)
Monocaprilocaprat de glicerol tip I (E471)
Dioxid de titan (E171)
Talc (E553b)
Oxid galben de fer (E172)
Lazcluze 240 mg comprimate filmate
Copolimer grefat de alcool polivinilic și macrogol (E1209)
Alcool polivinilic (E1203)
Monocaprilocaprat de glicerol tip I (E471)
Dioxid de titan (E171)
Talc (E553b)
Oxid roșu de fer (E172)
Oxid negru de fer (E172)
6.2 Incompatibilități
Nu este cazul.
6.3 Perioada de valabilitate
2 ani
6.4 Precauții speciale pentru păstrare
Acest medicament nu necesită condiții speciale de păstrare.
6.5 Natura și conținutul ambalajului
Lazcluze 80 mg comprimate filmate
Ambalaj cu blistere
Peliculă din policlorură de vinil-policlorotrifluoroetilenă (PVC-PCTFE) și folie de aluminiu perforabilă.
- O cutie conține 56 comprimate filmate (2 ambalaje de tip portofel, conținând 28 comprimate fiecare).
Flacon
Flacon alb, opac, din polietilenă de înaltă densitate (PEÎD), cu sistem de închidere din polipropilenă securizat pentru copii, care conține 60 sau 90 de comprimate. Fiecare cutie conține un flacon.
Lazcluze 240 mg comprimate filmate
Ambalaj cu blistere
Peliculă din policlorură de vinil-policlorotrifluoroetilenă (PVC-PCTFE) și folie de aluminiu perforabilă.
- O cutie conține 14 comprimate filmate (1 ambalaj de tip portofel conținând 14 comprimate).
- O cutie conține 28 comprimate filmate (2 ambalaje de tip portofel, conținând 14 comprimate fiecare).
Flacon
Flacon alb, opac, din polietilenă de înaltă densitate (PEÎD), cu sistem de închidere din polipropilenă securizat pentru copii, care conține 30 de comprimate filmate. Fiecare cutie conține un flacon.
Este posibil ca nu toate mărimile de ambalaj să fie comercializate.
6.6 Precauții speciale pentru eliminarea reziduurilor
Orice medicament neutilizat sau material rezidual trebuie eliminat în conformitate cu cerințele locale.
7. DEȚINĂTORUL AUTORIZAȚIEI DE PUNERE PE PIAȚĂ
Janssen-Cilag International NV
Turnhoutseweg 30
B-2340 Beerse
Belgia
8. NUMĂRUL(ELE) AUTORIZAȚIEI DE PUNERE PE PIAȚĂ
EU/1/24/1886/001
EU/1/24/1886/002
EU/1/24/1886/003
EU/1/24/1886/004
EU/1/24/1886/005
EU/1/24/1886/006
9. DATA PRIMEI AUTORIZĂRI SAU A REÎNNOIRII AUTORIZAȚIEI
10. DATA REVIZUIRII TEXTULUI
Informații detaliate privind acest medicament sunt disponibile pe site-ul Agenției Europene pentru Medicamente https://www.ema.europa.eu/.