TEVIMBRA 100 mg
Rezumatul caracteristicilor produsului (RCP)
- 1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI
- 2. COMPOZIȚIA CALITATIVĂ ȘI CANTITATIVĂ
- 3. FORMA FARMACEUTICĂ
-
4. DATE CLINICE
- 4.1 Indicații terapeutice
- 4.2 Doze și mod de administrare
- 4.3 Contraindicații
- 4.4 Atenționări și precauții speciale pentru utilizare
- 4.5 Interacțiuni cu alte medicamente și alte forme de interacțiune
- 4.6 Fertilitatea, sarcina și alăptarea
- 4.7 Efecte asupra capacității de a conduce vehicule și de a folosi utilaje
- 4.8 Reacții adverse
- 4.9 Supradozaj
- 5. PROPRIETĂȚI FARMACOLOGICE
1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI
Tevimbra 100 mg concentrat pentru soluție perfuzabilă.
2. COMPOZIȚIA CALITATIVĂ ȘI CANTITATIVĂ
Fiecare ml de concentrat pentru soluție perfuzabilă conține tislelizumab 10 mg.
Fiecare flacon de 10 ml conține tislelizumab 100 mg (100 mg/10 ml).
Tislelizumab este un anticorp monoclonal umanizat de tip imunoglobulină G4 (IgG4), modificat la nivelul segmentului Fc, produs prin tehnologia AND-ului recombinant în celule ovariene de hamster chinezesc.
Excipient cu efect cunoscut
Fiecare ml de concentrat pentru soluție perfuzabilă conține sodiu 1,6 mg și 0,2 mg polisorbat 20 (E432).
Pentru lista tuturor excipienților, vezi pct. 6.1.
3. FORMA FARMACEUTICĂ
Concentrat pentru soluție perfuzabilă (concentrat steril).
Soluție limpede până la ușor opalescentă, incoloră până la ușor gălbuie.
Soluția are un pH de aproximativ 6,5 și o osmolalitate de aproximativ 270 până la 330 mOsm/kg.
4. DATE CLINICE
4.1 Indicații terapeutice
Cancer bronho-pulmonar altul decât cel cu celule mici (NSCLC)
Tevimbra este indicat în asociere cu chimioterapie care conține săruri de platină ca tratament neoadjuvant, iar apoi continuat în monoterapie ca tratament adjuvant, pentru tratamentul pacienților adulți cu NSCLC rezecabil cu risc crescut de recurență (pentru criteriile de selecție, vezi pct. 5.1)
Tevimbra în asociere cu pemetrexed și chimioterapie pe bază de platină este indicat pentru tratamentul de primă intenție la pacienții adulți cu NSCLC, non-scuamos, ale căror tumori au expresie PD-L1 pe ≥50% din celulele tumorale), care nu prezintă EGFR sau ALK și care prezintă:
- NSCLC în stadiu local avansat și care nu sunt eligibili pentru rezecție chirurgicală sau chimioradioterapie pe bază de platină sau
- NSCLC în stadiu metastatic.
Tevimbra în asociere cu carboplatină și paclitaxel sau nab-paclitaxel este indicat pentru tratamentul de primă intenție la pacienții adulți cu NSCLC, scuamos, care prezintă:
- NSCLC în stadiu local avansat și care nu sunt eligibili pentru rezecție chirurgicală sau chimioradioterapie pe bază de platină sau
- NSCLC în stadiu metastatic.
Tevimbra în monoterapie este indicat pentru tratamentul pacienților adulți cu NSCLC în stadiu local avansat sau metastatic, după administrarea prealabilă de terapie pe bază de platină. Pacienților cu NSCLC cu EGFR mutant sau pozitiv pentru ALK trebuie, de asemenea, să li se fi administrat terapii țintite înainte de a li se administra tislelizumab.
Cancerul pulmonar cu celule mici (SCLC)
Tevimbra, în asociere cu chimioterapie cu etopozidă și platină, este indicat pentru tratamentul de primă linie al pacienților adulți cu SCLC în stadiu extins.
Adenocarcinom gastric sau de joncțiune esogastrică (G/JEG)
Tevimbra, în asociere cu chimioterapie pe bază de platină și fluoropirimidină, este indicat pentru tratamentul de primă linie al pacienților adulți cu adenocarcinom gastric sau de joncțiune esogastrică (G/JEG) avansat HER-2-negativ local avansat nerezecabil sau metastazat, ale căror tumori exprimă PD-L1 cu un scor de pozitivitate a zonei tumorale (TAP) ≥5% (vezi pct. 5.1).
Carcinom esofagian cu celule scuamoase (oesophageal squamous cell carcinoma - OSCC)
Tevimbra, în asociere cu chimioterapie pe bază de platină, este indicat pentru tratamentul de primă linie al pacienților adulți cu OSCC nerezecabil, în stadiu local avansat sau metastazat, ale căror tumori exprimă PD-L1 cu un scor TAP ≥5% (vezi pct. 5.1).
Tevimbra în monoterapie este indicat pentru tratamentul pacienților adulți cu OSCC nerezecabil, în stadiu local avansat sau metastazat, după administrarea prealabilă de chimioterapie pe bază de platină.
Carcinom nazofaringian (NPC)
Tevimbra, în asociere cu gemcitabină și cisplatină, este indicat pentru tratamentul de primă linie la pacienți adulți cu NPC recurent, refractar la intervenția chirurgicală curativă sau cu radioterapie, sau metastatic.
4.2 Doze și mod de administrare
Tratamentul cu Tevimbra trebuie să fie inițiat și supravegheat de către medici cu experiență în tratamentul neoplaziilor.
Testarea PD-L1
Dacă este specificat în indicație, selecția pacienților pentru tratamentul cu Tevimbra pe baza expresiei tumorale a PD-L1 trebuie evaluată printr-o testare cu un dispozitiv medical pentru diagnostic in vitro cu marcaj CE adecvat scopului testării. Dacă un dispozitiv medical pentru diagnostic in vitro cu marcaj CE nu este disponibil trebuie utilizat o altă testare validată (vezi pct. 4.1, 4.4 și 5.1).
Doze
Tevimbra în monoterapie
Doza recomandată de Tevimbra este de 200 mg o dată la 3 săptămâni sau 400 mg o dată la 6 săptămâni, administrată prin perfuzie intravenoasă. Pentru NSCLC rezecabil, în faza de tratament adjuvant, doza recomandată de Tevimbra este de 400 mg, administrată prin perfuzie intravenoasă o dată la 6 săptămâni.
Terapia cu Tevimbra în asociere
Doza recomandată de Tevimbra este de 200 mg o dată la 3 săptămâni sau 400 mg o dată la 6 săptămâni, administrată prin perfuzie intravenoasă, în asociere cu chimioterapie.
Când Tevimbra și chimioterapia sunt administrate în aceeași zi, Tevimbra trebuie administrat înaintea chimioterapiei. Trebuie consultat Rezumatul caracteristicilor produsului (RCP) pentru dozele de medicament chimioterapic, precum și pentru recomandări de utilizare a corticosteroizilor ca și premedicație în prevenirea reacțiilor adverse asociate chimioterapiei.
Durata tratamentului
Pacienții trebuie tratați cu Tevimbra până la progresia bolii sau până la apariția unui nivel inacceptabil de toxicitate (vezi pct. 5.1).
Pentru tratamentul neoadjuvant și adjuvant al NSCLC rezecabil, pacienții trebuie tratați cu Tevimbra neoadjuvant (200 mg o dată la 3 săptămâni) în asociere cu chimioterapie timp de 3 sau 4 cicluri sau până la progresia bolii care face imposibilă intervenția chirurgicală definitivă sau până la apariția unui nivel inacceptabil de toxicitate, urmat de tratament adjuvant cu Tevimbra (400 mg o dată la 6 săptămâni) în monoterapie timp de până la 8 cicluri sau până la recurența bolii, apariția metastazelor sau a toxicității inacceptabile.
Întârzierea sau întreruperea administrării dozei (vezi și pct. 4.4)
Nu se recomandă scăderi dozei de Tevimbra administrat în monoterapie sau în asociere cu alte medicamente. Administrarea Tevimbra trebuie întreruptă temporar sau definitiv în funcție de siguranță și tolerabilitate, conform descrierii din Tabelul 1.
Instrucțiuni detaliate pentru gestionarea reacțiilor adverse asociate sistemului imunitar sunt descrise la pct. 4.4.
Tabelul 1 Modificări recomandate ale tratamentului pentru Tevimbra
| Reacție adversă asociată sistemului imunitar | Gravitate1 | Modificarea tratamentului cu Tevimbra |
|---|---|---|
| Pneumonită | Grad 2 | Se întrerupe temporar tratamentul.2,3 |
| Grad 2 recidivant; grad 3 sau 4 | Tratamentul se întrerupe definitiv.3 | |
| Hepatită | ALT sau AST >3 până la 8 x LSN sau bilirubină totală >1,5 până la 3 x LSN | Se întrerupe temporar tratamentul.2,3 |
| ALT sau AST >8 x LSN sau bilirubină totală >3 x LSN | Tratamentul se întrerupe definitiv.3 | |
| Erupții cutanate tranzitorii | Grad 3 | Se întrerupe temporar tratamentul.2,3 |
| Grad 4 | Tratamentul se întrerupe definitiv.3 | |
| Reacții adverse cutanate grave (Severe cutaneous adverse reactions - SCARs) | SCARs suspectate, inclusiv SJS sau TEN | Se întrerupe temporar tratamentul.2,3 În caz de SJS sau TEN suspectate, nu se reia tratamentul dacă SJS/TEN nu au fost excluse, în consultare cu specialistul (specialiștii) adecvat(ți). |
| SCARs confirmate, inclusiv SJS sau TEN | Tratamentul se întrerupe definitiv. | |
| Colită | Grad 2 sau 3 | Se întrerupe temporar tratamentul.2,3 |
| Grad 3 recidivant; grad 4 | Tratamentul se întrerupe definitiv.3 | |
| Miozită/rabdomioliză | Grad 2 sau 3 | Se întrerupe temporar tratamentul.2,3 |
| Grad 3 recidivant; grad 4 | Tratamentul se întrerupe definitiv.3 | |
| Hipotiroidism | Grad 2, 3 sau 4 | Hipotiroidismul poate fi gestionat cu terapie de înlocuire, fără întreruperea tratamentului. |
| Hipertiroidism | Grad 3 sau 4 | Se întrerupe temporar tratamentul.2 Pentru grad 3 sau 4 care s-a ameliorat la grad ≤2 și este controlat cu terapie antitiroidiană, dacă este indicat, poate fi avută în vedere continuarea administrării Tevimbra după scăderea treptată a dozei de corticosteroid. În caz contrar, tratamentul trebuie întrerupt definitiv. |
| Insuficiență suprarenală | Grad 2 | Se are în vedere întreruperea temporară a tratamentului până la controlul prin HRT. |
| Grad 3 sau 4 | Se întrerupe temporar tratamentul.3 Pentru grad 3 sau 4 care s-a ameliorat la grad ≤2 și care este controlat cu HRT, dacă este indicat, poate fi avută în vedere continuarea administrării Tevimbra după scăderea treptată a dozei de corticosteroid. În caz contrar, tratamentul trebuie întrerupt definitiv.3 | |
| Hipofizită | Grad 2 | Se are în vedere întreruperea temporară a tratamentului până la controlul prin HRT. |
| Grad 3 sau 4 | Se întrerupe temporar tratamentul.2,3 Pentru grad 3 sau 4 care s-a ameliorat la grad ≤2 și care este controlat cu HRT, dacă este indicat, poate fi avută în vedere continuarea administrării Tevimbra după scăderea treptată a dozei de corticosteroid. În caz contrar, tratamentul trebuie întrerupt definitiv.3 | |
| Diabet zaharat de tip I | Diabet zaharat de tip I, asociat cu hiperglicemie de grad ≥3 (glucoză >250 mg/dl sau >13,9 mmol/l) sau cu cetoacidoză | Se întrerupe temporar tratamentul. Pentru grad 3 sau 4 care s-a ameliorat la grad ≤2 cu tratament cu insulină, dacă este indicat, poate fi avută în vedere continuarea administrării Tevimbra după ce se obține controlul metabolic. În caz contrar, tratamentul trebuie întrerupt definitiv. |
| Nefrită cu disfuncție renală | Grad 2 (creatinină >1,5 până la 3 x valoarea inițială sau >1,5 până la 3 x LSN) | Se întrerupe temporar tratamentul.2,3 |
| Grad 3 (creatinină >3 x valoarea inițială sau >3 până la 6 x LSN) sau grad 4 (creatinină >6 x LSN) | Tratamentul se întrerupe definitiv.3 | |
| Miocardită | Grad 2, 3 sau 4 | Tratamentul se întrerupe definitiv.3 |
| Toxicități neurologice | Grad 2 | Se întrerupe temporar tratamentul.2,3 |
| Grad 3 sau 4 | Tratamentul se întrerupe definitiv.3 | |
| Pancreatită | Pancreatită de grad 3 sau amilazemie de grad 3 sau 4 sau hiperlipazemie (>2 x LSN) | Se întrerupe temporar tratamentul.2,3 |
| Grad 4 | Tratamentul se întrerupe definitiv.3 | |
| Alte reacții adverse asociate sistemului imunitar | Grad 3 | Se întrerupe temporar tratamentul.2,3 |
| Grad 3 recidivant; grad 4 | Tratamentul se întrerupe definitiv.3 | |
| Alte reacții adverse | ||
| Reacții asociate perfuzării | Grad 1 | Se are în vedere administrarea preliminară a medicației în scopul prevenirii reacțiilor ulterioare asociate perfuzării. Se încetinește rata administrării perfuziei cu 50%. |
| Grad 2 | Se întrerupe perfuzarea. Se reia perfuzarea dacă reacțiile dispar sau se reduc până la gradul 1 și se încetinește rata administrării perfuziei cu 50%. | |
| Grad 3 sau 4 | Tratamentul se întrerupe definitiv. | |
ALT = alanin aminotransferază, AST = aspartat aminotransferază, HRT= terapie hormonală de substituție - hormone replacement therapy, SJS = sindrom Stevens-Johnson - Stevens Johnson syndrome, TEN = necroliză epidermică toxică - toxic epidermal necrolysis, LSN = limita superioară a normalului 1 Gradele de toxicitate sunt în conformitate cu Criteriile terminologice ale Institutului Național pentru Cancer privind reacțiile adverse Versiunea 4.0 (National Cancer Institute Common Terminology Criteria for Adverse Events – NCI-CTCAE, v4.0). Gradul hipofizitei este conform cu NCI-CTCAE v5.0. 2 Se reia tratamentul la pacienții cu rezolvare completă sau parțială (grad 0 până la 1) după reducerea treptată a dozei corticosteroizilor timp de cel puțin 1 lună. Tratamentul se întrerupe definitiv dacă nu există o rezoluție completă sau parțială în termen de 12 săptămâni de la inițierea corticosteroizilor sau nu se poate reduce doza de prednison la ≤10 mg/zi (sau echivalentul) în termen de 12 săptămâni de la inițierea corticosteroizilor. 3 Se recomandă doza inițială de prednison de 1 până la 2 mg/kg/zi sau echivalent, urmată de o scădere treptată până la ≤10 mg/zi (sau echivalentul) timp de cel puțin 1 lună, cu excepția pneumonitei, în cazul căreia se recomandă doza inițială de 2 până la 4 mg/kg/zi. | ||
Grupe speciale de pacienți
Copii și adolescenți
Siguranța și eficacitatea Tevimbra la pacienții cu vârsta sub 18 ani nu au fost încă stabilite. Nu sunt disponibile date.
Vârstnici
Nu este necesară ajustarea dozei la pacienții cu vârsta ≥65 ani (vezi pct. 4.8).
Insuficiență renală
Nu este necesară ajustarea dozei la pacienții cu insuficiență renală ușoară sau moderată. Datele de la pacienții cu insuficiență renală severă sunt prea limitate pentru a face recomandări de dozare pentru această populație (vezi pct. 5.2).
Insuficiență hepatică
Nu este necesară ajustarea dozei la pacienții cu insuficiență hepatică ușoară sau moderată. Datele de la pacienții cu insuficiență hepatică severă sunt prea limitate pentru a face recomandări de dozare pentru această populație (vezi pct. 5.2).
Mod de administrare
Tevimbra este indicat numai pentru utilizare intravenoasă. Se administrează sub formă de perfuzie și nu trebuie administrat sub formă de injecție intravenoasă accelerată sau injecție unică în bolus. Pentru instrucțiuni privind diluarea medicamentului înainte de administrare, vezi pct. 6.6.
Prima perfuzie de 200 mg trebuie administrată pe o perioadă de 60 minute. Dacă este bine tolerată, perfuziile ulterioare pot fi administrate pe o perioadă de 30 minute. Perfuzia trebuie administrată prin intermediul unei linii intravenoase care conține un filtru încorporat sau adăugat, steril, apirogen, cu un nivel scăzut de legare a proteinelor, de 0,2 sau 0,22 microni.
Perfuzia cu doza inițială de Tevimbra 400 mg trebuie administrată timp de 120 de minute (timp de 90 de minute dacă este utilizată ca tratament ulterior dozei de 200 mg o dată la 3 săptămâni). Dacă este bine tolerată, a doua perfuzie poate fi administrată timp de 60 de minute. Dacă a doua perfuzie este bine tolerată, perfuziile ulterioare pot fi administrate timp de 30 de minute.
Nu trebuie amestecate sau administrate concomitent prin aceeași linie de perfuzare alte medicamente.
4.3 Contraindicații
Hipersensibilitate la substanța activă sau la oricare dintre excipienții enumerați la pct. 6.1.
4.4 Atenționări și precauții speciale pentru utilizare
Trasabilitate
Pentru a avea sub control trasabilitatea medicamentelor biologice, numele și numărul lotului medicamentului administrat trebuie înregistrate cu atenție.
Evaluarea statusului PD-L1
Atunci când se evaluează statusul PD-L1 al tumorii este important să se aleagă o metodologie bine validată pentru a minimiza determinările fals negative sau fals pozitive.
Card pentru pacient
Pacienții tratați cu Tevimbra trebuie să primească Cardul pentru pacient pentru a fi informați cu privire la riscurile apariției reacțiilor adverse mediate imun pe durata terapiei cu Tevimbra (vezi și Prospectul).
Medicul prescriptor trebuie să discute cu pacientul riscurile apariției reacțiilor adverse mediate imun pe durata tratamentului cu Tevimbra.
Reacții adverse mediate imun
Au fost raportate reacții adverse mediate imun, inclusiv cazuri letale, pe durata tratamentului cu tislelizumab (vezi pct. 4.8). Cele mai multe dintre aceste evenimente s-au ameliorat odată cu întreruperea administrării tislelizumab, administrarea de corticosteroizi și/sau tratament de susținere. Au fost raportate reacții adverse mediate imun și după ultima doză de tislelizumab. Pot apărea simultan reacții adverse mediate imun care afectează mai mult de un sistem al corpului.
Pentru reacții adverse mediate imun suspectate, trebuie asigurată o evaluare adecvată pentru confirmarea etiologiei sau excluderea etiologiilor alternative, inclusiv a infecției. În funcție de gravitatea reacției adverse, administrarea tislelizumab trebuie să fie întreruptă și trebuie administrați corticosteroizi (vezi pct. 4.2). Pe baza datelor limitate din studii clinice, la pacienții ale căror reacții adverse mediate imun nu sunt controlate cu utilizarea corticosteroizilor, poate fi luată în considerare administrarea altor imunosupresoare sistemice (vezi pct. 4.2 și 4.8). După ameliorarea la grad ≤1, trebuie inițiată reducerea treptată a dozei de corticosteroizi și continuată pe o perioadă de cel puțin 1 lună.
La pacienții cu o boală autoimună (autoimmune disease - AID) preexistentă, datele din studiile observaționale sugerează că riscul de reacții adverse mediate imun după tratamentul cu inhibitori ai punctelor de control imunitar poate fi crescut comparativ cu riscul la pacienții fără AID preexistentă.
În plus, au fost frecvente exacerbările AID preexistente, însă majoritatea au fost ușoare și controlate terapeutic.
Pneumonită mediată imun
Pneumonita mediată imun, inclusiv cazuri letale, a fost raportată la pacienți cărora li s-a administrat tislelizumab. Pacienții trebuie monitorizați pentru semne și simptome ale pneumonitei. Pacienții suspectați de pneumonită trebuie evaluați prin imagistică radiografică, iar etiologiile infecțioase sau legate de boală trebuie excluse.
Pacienții cu pneumonită mediată imun trebuie tratați conform modificărilor tratamentului recomandate în Tabelul 1 (vezi pct. 4.2).
Hepatită mediată imun
Hepatita mediată imun, inclusiv cazuri letale, a fost raportată la pacienți cărora li s-a administrat tislelizumab. Pacienții trebuie monitorizați pentru semne și simptome ale hepatitei și modificărilor funcției hepatice. Trebuie efectuate analize ale funcției hepatice la momentul inițial și periodic pe durata tratamentului.
Pacienții cu hepatită mediată imun trebuie tratați conform modificărilor tratamentului recomandate în Tabelul 1 (vezi pct. 4.2).
Reacții cutanate mediate imun
Erupțiile cutanate tranzitorii sau dermatita au fost raportate la pacienți cărora li s-a administrat tislelizumab. Pacienții trebuie monitorizați pentru a se detecta apariția reacțiilor cutanate suspectate și trebuie excluse alte cauze. În funcție de gravitatea reacțiilor adverse cutanate, administrarea tislelizumab trebuie întreruptă temporar sau definitiv, conform recomandărilor din Tabelul 1 (vezi pct. 4.2).
Cazuri de reacții adverse cutanate grave (severe cutaneous adverse reactions - SCARs), inclusiv eritem multiform (EM), sindromul Stevens-Johnson (SJS) și necroliza epidermică toxică (TEN), unele dintre ele cu rezultat fatal au fost raportate la pacienți cărora li s-a administrat tislelizumab (vezi pct. 4.8). Pacienții trebuie monitorizați pentru semne sau simptome ale SCARs (de exemplu, sindrom prodromal al febrei, simptome similare gripei, leziuni ale mucoaselor sau erupții cutanate tranzitorii progresive) și trebuie excluse alte cauze. Pentru SCARs suspectate) trebuie să fie întreruptă temporar administrarea tislelizumab și pacientul trebuie să fie îndrumat către asistență specializată pentru evaluare și tratament. Dacă SCARsunt confirmate, administrarea tislelizumab trebuie întreruptă definitiv (vezi pct. 4.2).
Colită mediată imun
Colita mediată imun, frecvent asociată cu diaree, a fost raportată la pacienți tratați cu tislelizumab. Pacienții trebuie monitorizați pentru semne și simptome ale colitei. Trebuie excluse etiologiile infecțioase și legate de boală.
Pacienții cu colită mediată imun trebuie tratați conform modificărilor tratamentului recomandate în Tabelul 1 (vezi pct. 4.2).
Endocrinopatii mediate imun
Endocrinopatiile mediate imun, inclusiv tulburări tiroidiene, insuficiență suprarenală, hipofizită și diabet zaharat de tip I, au fost raportate la pacienți tratați cu tislelizumab. Acestea pot necesita tratament de susținere în funcție de tulburarea endocrină specifică. În cazul endocrinopatiilor mediate imun, poate fi necesară terapie de substituție hormonală pe termen lung (hormone replacement therapy
- HRT).
Pacienții cu endocrinopatii mediate imun trebuie tratați conform modificărilor tratamentului recomandate în Tabelul 1 (vezi pct. 4.2).
Tulburări tiroidiene
Tulburările tiroidiene, inclusiv tiroidită, hipotiroidism și hipertiroidism, au fost raportate la pacienți tratați cu tislelizumab. Pacienții trebuie monitorizați (la începutul tratamentului, periodic pe durata tratamentului și după cum este indicat pe baza evaluării clinice) pentru modificări ale funcției tiroidiene și semne și simptome clinice ale tulburărilor tiroidiene. Hipotiroidismul poate fi tratat cu HRT, fără întreruperea tratamentului și fără administrare de corticosteroizi. Hipertiroidismul poate fi tratat simptomatic (vezi pct. 4.2).
Insuficiență suprarenală
Insuficiența suprarenală a fost raportată la pacienți tratați cu tislelizumab Pacienții trebuie monitorizați pentru semne și simptome ale insuficienței suprarenale. Trebuie luată în considerare monitorizarea funcției suprarenale și a nivelurilor hormonale. Corticosteroizii și HRT trebuie administrați conform indicațiilor clinice (vezi pct. 4.2).
Hipofizită
Hipofizita a fost raportată la pacienți tratați cu tislelizumab. Pacienții trebuie monitorizați pentru semne și simptome ale hipofizitei/hipopituitarismului. Trebuie luată în considerare monitorizarea funcției hipofizei și a nivelurilor hormonale. Trebuie administrați corticosteroizi și HRT conform indicațiilor clinice (vezi pct. 4.2).
Diabet zaharat de tip I
Diabetul zaharat de tip I, inclusiv cetoacidoza diabetică, a fost raportat la pacienți tratați cu tislelizumab. Pacienții trebuie monitorizați pentru hiperglicemie și alte semne și simptome ale diabetului. Pentru diabet de tip I trebuie administrată insulină. La pacienții cu hiperglicemie severă sau cetoacidoză (grad ≥3), administrarea tislelizumab trebuie întreruptă temporar și trebuie administrat tratament antihiperglicemic (vezi pct. 4.2). Tratamentul cu tislelizumab poate fi reluat atunci când se obține controlul metabolic.
Nefrită mediată imun asociată cu disfuncție renală
Nefrita mediată imun asociată cu disfuncție renală a fost raportată la pacienți tratați cu tislelizumab. Pacienții trebuie monitorizați pentru modificări ale funcției renale (creșterea creatininemiei plasmatice) și trebuie excluse alte cauze ale disfuncției renale.
Pacienții cu nefrită mediată imun asociată cu disfuncție renală trebuie tratați conform modificărilor tratamentului recomandate în Tabelul 1 (vezi pct. 4.2).
Alte reacții adverse mediate imun
Au fost raportate și alte reacții adverse mediate imun semnificative clinic, asociate cu administrarea tislelizumab: miozită, miocardită, artrită, polimialgie reumatică, pericardită, cistită non-infecțioasă, trombocitopenie imună, encefalită, miastenia gravis, sindrom Sjögren și sindrom Guillain-Barré (vezi pct. 4.8).
Pacienții cu alte reacții adverse mediate imun trebuie tratați conform modificărilor de tratament recomandate în Tabelul 1 (vezi pct. 4.2).
Rejetul transplantului de organ solid
După punerea pe piață, la pacienți tratați cu inhibitori PD-1, a fost raportat rejetul transplantului de organ solid. Tratamentul cu tislelizumab poate crește riscul de rejet la pacienții cu transplant de organ solid. La acești pacienți trebuie evaluat beneficiul tratamentului cu tislelizumab față de riscul unui posibil rejet de organ.
Limfohistiocitoză hemofagocitară
Limfohistiocitoza hemofagocitară (hemophagocytic lymphohistiocytosis - HLH) a fost raportată la pacienții cărora li s-a administrat tislelizumab (vezi pct. 4.8). HLH este un sindrom care pune viața în pericol, caracterizat prin febră, erupție cutanată, limfadenopatie, hepato- și/sau splenomegalie și citopenii. Pacienții trebuie monitorizați pentru semne și simptome clinice de HLH. Pentru suspiciunea de HLH, tislelizumab trebuie întrerupt până la stabilirea diagnosticului și inițierea tratamentului pentru
HLH. Dacă se confirmă HLH, administrarea de tislelizumab trebuie întreruptă.
Reacții legate de perfuzare
Reacții grave legate de perfuzare (grad 3 sau mai înalt) au fost raportate la pacienți cărora li s-a administrat tislelizumab (vezi pct. 4.8). După punerea pe piață au fost raportate cazuri de anafilaxie, care includ reacție anafilactică și șoc anafilactic. Pacienții trebuie monitorizați pentru semne și simptome ale reacțiilor legate de perfuzare.
Reacțiile legate de perfuzare trebuie tratate conform recomandărilor din Tabelul 1 (vezi pct. 4.2).
Pacienți excluși din studii clinice
Pacienții cu oricare dintre următoarele afecțiuni au fost excluși din studii clinice: scor inițial de performanță ECOG mai mare sau egal cu 2; metastaze cerebrale sau leptomeningeale active; boală autoimună activă sau antecedente de boală autoimună, care poate recidiva; orice afecțiune care a necesitat tratament sistemic cu corticosteroizi (>10 mg/zi prednison sau echivalent) sau cu alte imunosupresoare în decursul celor 14 zile anterioare administrării tratamentului din cadrul studiului; infecție HIV activă sau netratată; purtători netratați ai virusului hepatitei B sau hepatitei C; antecedente de boală pulmonară interstițială; administrarea de vaccinuri vii în decursul celor 14 zile anterioare administrării tratamentului din cadrul studiului; infecție care necesită terapie sistemică în decursul celor 14 zile anterioare administrării tratamentului din cadrul studiului; antecedente de hipersensibilitate gravă la un alt anticorp monoclonal. În absența datelor, tislelizumab trebuie utilizat cu prudență la aceste populații, după o evaluare atentă a potențialului beneficiu/risc, în funcție de pacient.
Pacienți cu regim hiposodat
Fiecare ml din acest medicament conține 0,069 mmol (sau 1,6 mg) sodiu. Acest medicament conține 16 mg sodiu per flacon de 10 ml, echivalent cu 0,8% din doza maximă zilnică recomandată de OMS de 2 g sodiu pentru un adult.
Tevimbra se diluează în soluție perfuzabilă de clorură de sodiu 9 mg/ml (0,9%). Acest lucru trebuie luat în considerare la pacienții care urmează o dietă cu aport controlat de sodiu (vezi pct. 6.6).
Polisorbat 20 (E432)
Acest medicament conține 0,2 mg de polisorbat 20 în fiecare ml de concentrat, ceea ce echivalează cu 4 mg în două flacoane de 10 ml dintr-o singură perfuzie de Tevimbra. Polisorbații pot cauza reacții alergice. Pacienții cu alergii cunoscute trebuie luați în considerare.
4.5 Interacțiuni cu alte medicamente și alte forme de interacțiune
Tislelizumab este un anticorp monoclonal umanizat, eliminat din circulație prin catabolism. Drept urmare, nu au fost efectuate studii formale de interacțiune farmacocinetică. Deoarece anticorpii monoclonali nu sunt metabolizați de enzimele citocromului P450 (CYP) sau de alte enzime care metabolizează medicamentele, nu se anticipează că inhibarea sau inducerea acestor enzime de către medicamente administrate concomitent va afecta farmacocinetica tislelizumab.
Utilizarea corticosteroizilor sistemici și a altor imunosupresoare la momentul inițial, înainte de începerea administrării tislelizumab, cu excepția dozelor scăzute de corticosteroizi sistemici (10 mg/zi prednison sau echivalent), trebuie evitată din cauza posibilei interferențe cu activitatea farmacodinamică și eficacitatea tislelizumabului. Cu toate acestea, corticosteroizii sistemici și alte imunosupresoare pot fi utilizați după începerea administrării tislelizumab pentru a trata reacțiile adverse mediate imun (vezi pct. 4.4). Corticosteroizii pot fi utilizați și ca premedicație atunci când tislelizumab este utilizat în asociere cu chimioterapie, ca profilaxie antiemetică și/sau pentru a atenua reacțiile adverse asociate chimioterapiei.
4.6 Fertilitatea, sarcina și alăptarea
Femei aflate la vârsta fertilă/Contracepție
Tislelizumab nu trebuie utilizat la femeile aflate la vârsta fertilă care nu utilizează metode contraceptive eficace, cu excepția cazului în care starea clinică a femeii necesită tratament cu tislelizumab. Femeile aflate la vârsta fertilă trebuie să utilizeze metode contraceptive eficace (metode care au ca rezultat rate de sarcină mai mici de 1%) pe durata tratamentului și timp de cel puțin 4 luni după ultima doză de tislelizumab.
Sarcina
Nu există date disponibile privind utilizarea tislelizumab la femeile gravide. Pe baza mecanismului său de acțiune, tislelizumab poate dăuna fătului atunci când este administrat la femei însărcinate.
Nu au fost efectuate studii privind reproducerea la animale cu tislelizumab. Cu toate acestea, în cazul modelelor murine de sarcină, blocarea semnalizării PD-1/PD-L1 s-a dovedit a perturba toleranța la făt și a conduce la rate crescute ale pierderii sarcinilor.
Se cunoaște că IgG4 (imunoglobulinele) umane traversează bariera placentară. Prin urmare, fiind o variantă a IgG4, tislelizumab are potențialul de a fi transmis de la mamă la fătul în creștere. Femeile trebuie să fie informate cu privire la riscul potențial pentru un făt.
Tislelizumab nu trebuie utilizat în timpul sarcinii, cu excepţia cazului în care starea clinică a femeii impune tratament cu tislelizumab.
Alăptarea
Nu se cunoaşte dacă tislelizumab se excretă în laptele uman. De asemenea, sunt necunoscute efectele sale asupra nou-născuţilor/sugarilor alăptați și asupra secreției de lapte.
Dat fiind potențialul apariției reacțiilor adverse grave la Tevimbra la nou-născuți/sugari alăptați, femeilor trebuie să li se recomande să nu alăpteze în timpul tratamentului și timp de minimum 4 luni de la administrarea ultimei doze de Tevimbra.
Fertilitatea
Nu sunt disponibile date clinice cu privire la efectele posibile ale tislelizumab asupra fertilității. Nu au fost efectuate studii privind toxicitatea asupra funcției de reproducere și asupra dezvoltării cu tislelizumab. Pe baza unui studiu privind toxicitatea la doze repetate, cu durata de 3 luni, nu au existat efecte notabile asupra organelor reproducătoare masculine și feminine la maimuța cynomolgus, atunci când tislelizumab a fost administrat în doze de 3, 10 sau 30 mg/kg la interval de 2 săptămâni, timp de 13 săptămâni (7 administrări de doze) (vezi pct. 5.3).
4.7 Efecte asupra capacității de a conduce vehicule și de a folosi utilaje
Tevimbra are influență mică asupra capacității de a conduce vehicule sau de a folosi utilaje. La unii pacienți a fost raportată fatigabilitate după administrarea tislelizumab (vezi pct. 4.8).
4.8 Reacții adverse
Rezumatul profilului de siguranță
Siguranța tislelizumab în monoterapie se bazează pe date cumulate de la 1952 pacienți, cu mai multe tipuri de tumori, cărora li s-a administrat tislelizumab 200 mg la fiecare 3 săptămâni. Cele mai frecvente reacții adverse (≥ 20%) au fost anemia (27,7%), creșterea valorilor aspartat aminotransferazei (24,7%), oboseală (24,6%) și creșterea valorilor alanin aminotransferazei (22,0%). Cele mai frecvente reacții adverse de grad 3/4 (≥ 2%) au fost anemia (4,8%), creșterea valorilor aspartat aminotransferazei (3,7%), pneumonia (3,6%), hiponatremia (2,9%), creșterea valorilor bilirubinei sanguine (2,8%), hipertensiunea (2,4%) și fatigabilitatea (2,1%). Dintre pacienți 1,0% au prezentat reacții adverse care au condus la deces. Reacțiile adverse care au condus la deces au fost pneumonia (0,61%), pneumonita (0,10%), hepatita (0,10%), trombocitopenia (0,05%), dispneea (0,05%) și apetitul alimentar scăzut (0,05%). Dintre cei 1.952 pacienți, 40,7% au fost expuși la tislelizumab timp de mai mult de 6 luni, iar 24,7% au fost expuși timp de mai mult de 12 luni.
Siguranța tislelizumab administrat în asociere cu chimioterapie se bazează pe datele de la 1950 de pacienți cu mai multe tipuri de tumori care au primit 200 mg tislelizumab la fiecare 3 săptămâni, cu cu excepția studiului BGB A317-315, în care pacienții au primit și tislelizumab în doză de 400 mg o dată la 6 săptămâni ca tratament adjuvant după terapia neoadjuvantă și intervenția chirurgicală. Cele mai frecvente reacții adverse (≥ 20%) au fost neutropenia (71,6%), anemia (67,2%), trombocitopenia (48,7%), greața (43,3%), fatigabilitatea (40,8%), apetitul alimentar scăzut (40,1%), creșterea valorilor aspartat aminotransferazei (30,6%), valori crescute ale alanin aminotransferazei (30,3%), erupția cutanată (21,4%) și diareea (20,3%). Cele mai frecvente reacții adverse de grad 3/4 (≥ 2%) au fost neutropenia (45,2%), anemia (14,5%), trombocitopenia (14,1%), hiponatremia (4,6%), hipokaliemia (4,5%), fatigabilitatea (4,2%), pneumonia (4,0%), limfopenia (3,1%), erupții cutanate tranzitorii (2,9%), apetit scăzut (2,6%), valori crescute ale aspartat aminotransferazei (2,2%), valori crescute ale alanin aminotransferazei (2,1%). 1,3% dintre pacienți au prezentat reacții adverse finalizate cu deces. Reacțiile adverse care au dus la deces au fost pneumonia (0,50%), pneumonita (0,30%), dispneea (0,20%), miocardita (0,20%), hepatita (0,05%), trombocitopenia (0,05%), colita (0,05%), hipokaliemia (0,05%) și miozita (0,05%). Dintre cei 1950 de pacienți, 56,5% au fost expuși la tislelizumab timp de 6 luni sau mai mult, iar 31,9% au fost expuși timp de 12 luni sau mai mult.
Listă tabelară a reacțiilor adverse
Reacțiile adverse raportate în setul de date centralizat pentru pacienții tratați cu Tevimbra în monoterapie (N= 1952) și în asociere cu chimioterapie (N = 1950) sunt prezentate în Tabelul 2. Reacțiile adverse sunt enumerate conform bazei de date MedDRA pe aparate sisteme și organe. În cadrul fiecărei clase de aparate, sisteme și organe, reacțiile adverse sunt prezentate în ordinea descrescătoare a frecvenței. Categoria de frecvență corespunzătoare pentru fiecare reacție adversă este definită ca: foarte frecvente (≥1/10); frecvente (≥1/100 și <1/10); mai puțin frecvente (≥1/1 000 și <1/100); rare (≥1/10 000 și <1/1 000); foarte rare (<1/10 000); cu frecvență necunoscută (care nu poate fi estimată din datele disponibile). În cadrul fiecărei grupe de frecvență, reacțiile adverse sunt prezentate în ordinea descrescătoare a gravității.
Tabelul 2 Reacții adverse la administrarea Tevimbra în monoterapie (N = 1952) și în asociere cu chimioterapie (N = 1950)
| Tislelizumab în monoterapie N = 1952 | Tislelizumab plus chimioterapie N = 1950 | |
| Reacții adverse | Categoria de frecvență (Toate gradele) | Categoria de frecvență (Toate gradele) |
| Infecții și infestări | ||
|---|---|---|
| Pneumonie1 | Frecvente* | Foarte frecvente* |
| Tulburări hematologice și limfatice | ||
| Anemie2 | Foarte frecvente | Foarte frecvente |
| Trombocitopenie3 | Foarte frecvente* | Foarte frecvente* |
| Neutropenie4 | Frecvente | Foarte frecvente |
| Limfopenie5 | Frecvente | Foarte frecvente |
| Limfohistiocitoză hemofagocitară | Cu frecvență necunoscută | Rare |
| Tulburări ale sistemului imunitar | ||
| Sindromul Sjögren | # | Mai puțin frecvente |
| Tulburări endocrine | ||
| Hipotiroidism6 | Foarte frecvente | Foarte frecvente |
| Hipertiroidism7 | Frecvente | Frecvente |
| Tiroidită8 | Frecvente | Mai puțin frecvente |
| Insuficiență suprarenală9 | Mai puțin frecvente | Mai puțin frecvente |
| Hipofizită10 | Mai puțin frecvente | Mai puțin frecvente |
| Tulburări metabolice şi de nutriţie | ||
| Hiperglicemie11 | Frecvente | Foarte frecvente |
| Hiponatremie12 | Frecvente | Foarte frecvente |
| Hipokalemie13 | Frecvente | Foarte frecvente* |
| Diabet zaharat14 | Mai puțin frecvente | Frecvente |
| Tulburări ale sistemului nervos | ||
| Sindrom Guillain-Barré | Rare | Rare |
| Encefalită15 | # | Rare |
| Miastenia gravis | # | Rare |
| Tulburări oculare | ||
| Uveită16 | Mai puțin frecvente | Mai puțin frecvente |
| Tulburări cardiace | ||
| Miocardită17 | Mai puțin frecvente | Frecvente* |
| Pericardită | Mai puțin frecvente | Rare |
| Tulburări vasculare | ||
| Hipertensiune arterială18 | Frecvente | Frecvente |
| Tulburări respiratorii, toracice şi mediastinale | ||
| Tuse | Foarte frecvente | Foarte frecvente |
| Dispnee | Frecvente* | Frecvente* |
| Pneumonită19 | Frecvente* | Frecvente* |
| Tulburări gastro-intestinale | ||
| Greață | Foarte frecvente | Foarte frecvente |
| Diaree20 | Foarte frecvente | Foarte frecvente |
| Stomatită21 | Frecvente | Frecvente |
| Pancreatită22 | Mai puțin frecvente | Frecvente |
| Colită23 | Mai puțin frecvente | Frecvente* |
| Boală celiacă | Rare | # |
| Tulburări hepatobiliare | ||
| Hepatită24 | Frecvente* | Frecvente* |
| Afecţiuni cutanate şi ale ţesutului subcutanat | ||
| Erupții cutanate tranzitorii25 | Foarte frecvente | Foarte frecvente |
| Prurit | Foarte frecvente | Foarte frecvente |
| Vitiligo26 | Mai puțin frecvente | Mai puțin frecvente |
| Eritem multiform | Mai puțin frecvente | Rare |
| Sindrom Stevens-Johnson | Rare | # |
| Necroliză epidermică toxică27 | Necunoscute* | Necunoscute* |
| Tulburări musculo-scheletice şi ale ţesutului conjunctiv | ||
| Artralgie | Frecvente | Foarte frecvente |
| Mialgie | Frecvente | Frecvente |
| Miozită28 | Mai puțin frecvente | Mai puțin frecvente* |
| Artrită29 | Mai puțin frecvente | Frecvente |
| Tulburări renale şi ale căilor urinare | ||
| Nefrită30 | Mai puțin frecvente | Mai puțin frecvente |
| Cistită non-infecțioasă31 | Rare | # |
| Tulburări generale şi la nivelul locului de administrare | ||
| Fatigabilitate32 | Foarte frecvente | Foarte frecvente |
| Febră33 | Foarte frecvente | Foarte frecvente |
| Apetit alimentar scăzut | Foarte frecvente* | Foarte frecvente |
| Investigaţii diagnostice | ||
| Valori crescute ale aspartat aminotransferazei | Foarte frecvente | Foarte frecvente |
| Valori crescute ale alanin aminotransferazei | Foarte frecvente | Foarte frecvente |
| Valori crescute ale bilirubinemiei34 | Foarte frecvente | Foarte frecvente |
| Valori crescute ale forfatazei alcaline în sânge | Frecvente | Frecvente |
| Valori crescute ale creatininei în sânge | Frecvente | Foarte frecvente |
| Leziuni, intoxicaţii şi complicaţii legate de procedurile utilizate | ||
| Reacție asociată perfuzării35 | Frecvente | Frecvente |
1 Pneumonia include termenii agreați (preferred terms - PTs) de pneumonie, infecție a tractului respirator inferior, infecție bacteriană a tractului respirator inferior, pneumonie bacteriană, pneumonie fungică, pneumonie cu Pneumocystis jirovecii, aspergiloza bronhopulmonară, pneumonia cu candida, pneumonia micoplasmatică, pneumonia stafilococică și pneumonia virală. 2 Anemia include termenii agreați de anemie și scăderea valorii hemoglobinei. 3 Trombocitopenia include termenii agreați de trombocitopenie, scăderea numărului de plachete sanguine și trombocitopenie imună. 4 Neutropenia include termenii agreați de neutropenie și scăderea numărului de neutrofile. 5 Limfopenia include termenii agreați de limfopenie, scăderea numărului de limfocite și scăderea procentului de limfocite. 6 Hipotiroidismul include termenii agreați de hipotiroidism, valori crescute ale anticorpilor antitiroidieni, hipotiroidism mediat imun, valori scăzute ale hormonilor tiroidieni, scăderea valorii tiroxinei libere, scăderea valorii tri-iodotironinei libere, scăderea valorii tri-iodotironinei, hipotiroidism primar, hipotiroidism central și scăderea valorii tiroxinei. 7 Hipertiroidismul include termenii agreați de hipertiroidism, hipertiroidism, hipertiroidism mediat imun, creșterea valorii tiroxinei libere, creșterea valorii tiroxinei, creșterea valorii tri-iodotironinei libere și creșterea valorii tri-iodotironinei. 8 Tiroidita include termenii agreați de tiroidită, tiroidită autoimună, tiroidita imunomediată, tiroidita silențioasă și tiroidită subacută. 9 Insuficiența suprarenală include termenii agreați de boală Addison, insuficiență suprarenală, deficit de glucocorticoizi, insuficiență suprarenală mediată imun, insuficiență adrenocorticală primară și insuficiență adrenocorticală secundară. 10 Hipofizita include termenii agreați de hipofizită și hipopituitarism. 11 Hiperglicemia include termenii agreați de hiperglicemie și creșterea valorilor glucozei din sânge. 12 Hiponatremia include termenii agreați de hiponatremie și scăderea valorilor sodiului din sânge. 13 Hipokalemia include termenii agreați de hipokalemie și scăderea valorilor potasiului din sânge. 14 Diabetul zaharat include termenii agreați de diabet zaharat, cetoacidoză diabetică, cetoză diabetică, cetoacidoză, diabet zaharat de tip I și diabet autoimun latent la adulți. 15 Encefalita include termenul agreat de encefalita mediată imun. 16 Uveita include termenii agreați de corioretinită, iridociclită, uveită și irită. 17 Miocardita include termenii agreați de miocardită, miocardită mediată imun și miocardită autoimună. 18 Hipertensiunea include termenii agreați de hipertensiune, creșterea tensiunii arteriale și hipertensiune esențială. 19 Pneumonita include termenii agreați de pneumonită, boală pulmonară mediată imun, boală pulmonară interstițială și pneumonită organizantă. 20 Diareea include termenii agreați de diaree și scaune frecvente. 21 Stomatita include termenii agreați de stomatită, ulcerații la nivelul gurii, eroziunea mucoasei orale și ulcer aftos. 22 Pancreatita include termenii agreați de amilazemie crescută, lipazemie crescută, pancreatită și pancreatită acută. 23 Colita include termenii agreați de colită autoimună, colită, colită ulcerativă și enterocolită mediată imun. 24 Hepatita include termenii agreați de hepatită, leziuni hepatice induse de medicamente, hepatotoxicitate, funcție hepatică anormală, hepatită mediată imun, leziuni hepatice și hepatită autoimună. 25 Erupțiile cutanate tranzitorii includ termenii agreați de erupții cutanate tranzitorii, erupții cutanate tranzitorii maculopapulare, eczeme, erupții cutanate tranzitorii eritematoase, dermatită, dermatoză neutrofilă febrilă acută, dermatită autoimună, dermatită alergică, dermatită exfoliativă, erupții cutanate tranzitorii papulare, urticarie, eritem, exfolierea pielii, erupții cutanate tranzitorii maculare, psoriazis, erupții cutanate tranzitorii pustulare, dermatită acneiformă, erupții cutanate tranzitorii pruritice, keratoză lichenoidă, dermatita mâinii, dermatită mediată imun, erupții cutanate tranzitorii foliculare, eritem nodos și | ||
pemfigoid.
26 Vitiligo include termenii agreați de depigmentare cutanată leucodermică, depigmentare cutanată și vitiligo. 27 Experiența după punerea pe piață.
28 Miozita include termenii agreați de miozită, rabdomioliză și miozită mediată imun.
29 Artrita include termenii agreați de artrită, poliartrită și artrită mediată imun și poliartrită.
30 Nefrita include termenii agreați de nefrită, glomeruloscleroză segmentară focală, glomerulonefrită membranoasă, tulburare renală mediată imun, nefrită tubulointerstițială și nefrită mediată imun. 31 Cistita non-infecțioasă include termenii agreați de cistită non-infecțioasă și cistită mediată imun. Cazurile de cistită mediată imun au fost raportate după punerea pe piață.
32 Fatigabilitatea include termenii agreați de fatigabilitate, astenie, stare de rău, decondiționare fizică și letargie.
33 Febră include termenii agreați de temperatură corporală crescută și febră.
34 Valorile crescute ale bilirubinemiei includ termenii agreați de valori crescute ale bilirubinemiei, valori crescute ale bilirubinemiei conjugate, valori crescute ale bilirubinei neconjugate și hiperbilirubinemia. 35 Reacția asociată perfuzării include termenii agreați de reacție anafilactică, frisoane, edem cornean, dermatică alergică, erupție la medicament, hipersensibilitate la medicament, edem al feței, umflare a gingiilor, hipersensibilitate, obstrucție laringiană, edem laringian, edem al buzelor, umflare a buzelor, umflare a gurii, prurit alergic, erupție cutanată, erupție cutanată eritematoasă, erupție maculară, erupție pruriginoasă, rinită alergică, umflarea feței, edem al limbii, hipersensibilitate de tip I, urticarie, reacție legată de perfuzie și reacție de hipersensibilitate legată de perfuzie.
* Inclusiv rezultate letale
# Neraportat în această setare grupată
Descrierea reacțiilor adverse selectate
Datele de mai jos reflectă informațiile privind reacțiile adverse semnificative la medicament pentru tislelizumab în monoterapie, administrat în cadrul studiilor clinice. Detaliile reacțiilor adverse semnificative pentru tislelizumab administrat în asociere cu chimioterapie sunt prezentate dacă apar diferențe semnificative clinic în comparație cu tislelizumab în monoterapie.
Pneumonită mediată imun
La pacienții tratați cu tislelizumab în monoterapie, pneumonita mediată imun a apărut la 5,1% dintre pacienți, inclusiv evenimente de grad 1 (1,3%), grad 2 (2,1%), grad 3 (1,3%), grad 4 (0,3%) și grad 5 (0,1%).
Durata mediană de la prima doză până la debutul evenimentului a fost de 4,1 luni (interval: 1,0 zile până la 55,0 luni), iar durata mediană de la debut până la rezolvare a fost de 2,8 luni (interval: 7,0+ zile până la 33,7+ luni). Tislelizumab a fost întrerupt definitiv la 1,8% dintre pacienți iar tratamentul cu tislelizumab a fost întrerupt temporar la 1,9% dintre pacienți. Pneumonita s-a rezolvat la 47,0% dintre pacienți.
La pacienții tratați cu tislelizumab în monoterapie, pneumonita a apărut mai frecvent la pacienții cu un istoric de administrare anterioară a radioterapiei la nivel toracic (8,4%) decât la pacienții cărora nu li s-a administrat anterior radioterapie la nivel toracic (3,6%).
Pneumonita a apărut la 11,2% dintre pacienții cu NSCLC tratați cu tislelizumab în asociere cu chimioterapia. La pacienții cu NSCLC tratați cu tislelizumab în monoterapie, pneumonita apărut la 8,3% dintre pacienți.
Hepatită mediată imun
La pacienții tratați cu tislelizumab în monoterapie, hepatita mediată imun a apărut la 1,2% dintre pacienți, inclusiv evenimente de grad 1 (0,1%), grad 2 (0,2%), grad 3 (0,6%) și grad 4 (0,3%).
Durata mediană de la prima doză până la debutul evenimentului a fost de 22,0 zile (interval: 1,0 zile până la 4,1 luni), iar durata mediană de la debut până la rezolvare a fost de 1,1 luni (interval: 6,0 zile până la 6,6 luni). Tislelizumab a fost întrerupt definitiv la 0,3% dintre pacienți iar tratamentul cu tislelizumab a fost întrerupt temporar la 0,8% dintre pacienți pentru hepatită mediată imun. Hepatita sa rezolvat la 60,9% dintre pacienți.
Reacții adverse cutanate mediate imun
La pacienții tratați cu tislelizumab în monoterapie, reacțiile adverse cutanate mediate imun au apărut la 12,6% dintre pacienți, inclusiv evenimente de grad 1 (7,7%), grad 2 (3,7%), grad 3 (1,0%) și grad 4 (0,1%).
Durata mediană de la prima doză până la debutul evenimentului a fost de 1,5 luni (interval: 1,0 zile până la 36,1 luni). Durata mediană de la debut până la rezolvare a fost de 1,1 luni (interval: 1,0 zile până la 36,7 luni). Tislelizumab a fost întrerupt definitiv la 0,1% dintre pacienți iar tratamentul cu tislelizumab a fost întrerupt temporar la 1,3% dintre pacienți. Reacțiile adverse cutanate s-au rezolvat la 72,0% dintre pacienți.
Cazuri de SJS și TEN au fost raportate din experiența ulterioară punerii pe piață, unele cu rezultat fatal (vezi pct. 4.2 și 4.4).
Colită mediată imun
La pacienții tratați cu tislelizumab în monoterapie, colita mediată imun a apărut la 0,6% dintre pacienți, inclusiv evenimente de grad 2 (0,4%) și grad 3 (0,2%).
Durata mediană de la prima doză până la debutul evenimentului a fost de 6,0 luni (interval: 6,0 zile până la 26,5 luni), iar durata mediană de la debut până la rezolvare a fost de 28,0 zile (interval: 9,0 zile până la 26,7 luni). Tislelizumab nu a fost întrerupt definitiv la 0,1% dintre pacienți, iar tratamentul cu tislelizumab a fost întrerupt temporar la 0,4% dintre pacienți. Colita s-a rezolvat la 81,8% dintre pacienți.
Miozită mediată imun/rabdomioliză
La pacienții tratați cu tislelizumab în monoterapie, miozita mediată imun/rabdomioliza a apărut la 0,8% dintre pacienți, inclusiv evenimente de grad 1 (0,3%), grad 2 (0,3%), grad 3 (0,2%) și grad 4 (0,1%).
Durata mediană de la prima doză până la debutul evenimentului a fost de 1,5 luni (interval: 15,0 zile până la 39,3 luni), iar durata mediană de la debut până la rezolvare a fost de 1,2 luni (interval: 5,0 zile până la 5,2 luni). Tislelizumab a fost întrerupt definitiv la 0,2% dintre pacienți iar tratamentul cu tislelizumab a fost întrerupt temporar la 0,5% dintre pacienți. Miozita/rabdomioliza s-a rezolvat la 75,0% dintre pacienți.
Endocrinopatii mediate imun
Tulburări tiroidiene
Hipotiroidism:
La pacienții tratați cu tislelizumab în monoterapie, hipotiroidismul a apărut la 13,8% dintre pacienți, inclusiv evenimente de grad 1 (6,4%), grad 2 (7,3%), grad 3 (0,1%) și grad 4 (0,1%).
Durata mediană de la prima doză până la debutul evenimentului a fost de 4,0 luni (interval: 1,0 zile până la 29,9 luni). Durata mediană de la debut până la rezolvare a fost de 2,1 luni (interval: 2,0 zile până la 27,0 luni). Tislelizumab a fost întrerupt definitiv la 0,1% pacienți, iar tratamentul cu tislelizumab a fost întrerupt temporar la 0,6% dintre pacienți. Hipotiroidismul s-a rezolvat la 36,4% dintre pacienți.
Hipertiroidism:
La pacienții tratați cu tislelizumab în monoterapie, hipertiroidismul a apărut la 5,1% dintre pacienți, inclusiv evenimente de grad 1 (4,4%) și grad 2 (0,7%).
Durata mediană de la prima doză până la debutul evenimentului a fost de 2,1 luni (interval: 6,0 zile până la 39,4 luni). Durata mediană de la debut până la rezolvare a fost de 1,4 luni (interval: 8,0 zile până la 22,1 luni). Tislelizumab a fost întrerupt definitiv la 0,1% dintre pacienți iar tratamentul cu tislelizumab nu a fost întrerupt temporar la 0,3% dintre pacienți. Hipertiroidismul s-a rezolvat la 77,0% dintre pacienți.
Tiroidită:
La pacienții tratați cu tislelizumab în monoterapie, tiroidita a apărut la 1,1% dintre pacienți, inclusiv evenimente de grad 1 (0,5%) și grad 2 (0,6%).
Durata mediană de la prima doză până la debutul evenimentului a fost de 2,0 luni (interval: 14,0 zile până la 20,7 luni). Durata mediană de la debut până la rezolvare a fost de 2,0 luni (interval: 20,0 zile până la 15,3 luni). Tislelizumab nu a fost întrerupt definitiv la niciun pacient iar tratamentul cu tislelizumab a fost întrerupt temporar la 0,2% dintre pacienți. Tiroidita s-a rezolvat la 38,1% dintre pacienți.
Insuficiență suprarenală
La pacienții tratați cu tislelizumab în monoterapie, insuficiența suprarenală a apărut la 0,5% dintre pacienți, inclusiv evenimente de grad 2 (0,3%), grad 3 (0,2%) și grad 4 (0,1%).
Durata mediană de la prima doză până la debutul evenimentului a fost de 10,3 luni (interval: 1,4 luni până la 16,9 luni). Durata mediană de la debut până la rezolvare a fost de 1,9 luni (interval: 30 de zile până la 13,6 luni). Tislelizumab nu a fost întrerupt definitiv la niciun pacient iar tratamentul cu tislelizumab a fost întrerupt temporar la 0,4% dintre pacienți. Insuficiența suprarenală s-a rezolvat la 30,0% dintre pacienți.
Hipofizită
La pacienții tratați cu tislelizumab în monoterapie, hipofizita (grad 2) a apărut la 0,3% dintre pacienți.
Durata mediană de la prima doză până la debutul evenimentului a fost de 9,0 luni (interval: 22,0 zile până la 16,2 luni). Durata mediană de la debut până la remisie a fost de 2,3 luni (numai 1 singur eveniment remis). Tislelizumab nu a fost întrerupt definitiv la niciunul dintre pacienți, iar tratamentul cu tislelizumab nu a fost întrerupt la niciun pacient. Hipofizita s-a remis la 20,0 dintre pacienți.
Diabet zaharat de tip 1
La pacienții tratați cu tislelizumab în monoterapie, diabetul zaharat de tip 1 a apărut la 0,6% dintre pacienți, inclusiv evenimente de grad 1 (0,1%), grad 2 (0,3%), grad 3 (0,2%) și grad 4 (0,1%).
Durata mediană de la prima doză până la debutul evenimentului a fost de 6,5 luni (interval: 1,1 zile până la 36,1 luni). Durata mediană de la debut până la rezolvare a fost de 22,0 luni (interval: 5,0 zile până la 3,6 luni). Tislelizumab a fost întrerupt definitiv la 0,2% dintre pacienți iar tratamentul cu tislelizumab a fost întrerupt temporar la 0,2% dintre pacienți. Diabetul zaharat de tip 1 s-a rezolvat la 8,3% dintre pacienți.
Nefrită mediată imun și disfuncție renală
La pacienții tratați cu tislelizumab în monoterapie, nefrita mediată imun și disfuncția renală au apărut la 0,2% dintre pacienți, inclusiv evenimente de grad 1 (0,1%), grad 2 (0,1%) și grad 3 (0,1%).
Durata mediană de la prima doză până la debutul evenimentului a fost de 1,5 luni (interval: 15,0 zile până la 12,1 luni). Durata mediană de la debut până la rezolvare a fost 9,0 zile (aceeași pentru 2 evenimente remise). Tislelizumab a fost întrerupt definitiv la 0,1% dintre pacienți iar tratamentul cu tislelizumab a fost întrerupt temporar la 0,1% dintre pacienți. Nefrita mediată imun și disfuncția renală s-au rezolvat la 50,0% dintre pacienți.
Miocardită mediată imun
La pacienții tratați cu tislelizumab în monoterapie, miocardita mediată imun a apărut la 0,8% dintre pacienți, inclusiv evenimente de grad 1 (0,4%), grad 2 (0,2%), grad 3 (0,2%) și grad 4 (0,1%).
Durata mediană de la prima doză până la debutul evenimentului a fost de 1,6 luni (interval: 14,0 zile până la 33,6 luni), iar durata mediană de la debut până la rezolvare a fost de 1,2 luni (interval: 4,0 zile până la 15,6 luni). Tislelizumab a fost întrerupt definitiv la 0,4% dintre pacienți iar tratamentul cu tislelizumab a fost întrerupt temporar la 0,4% dintre pacienți. Miocardita s-a rezolvat la 60,0% dintre pacienți.
Miocardita a apărut la 1,2% dintre pacienții tratați cu tislelizumab în asociere cu chimioterapia, inclusiv evenimente de gradul 5 (0,2%).
Efecte asupra clasei de inhibitori ai punctelor de control imunitar
În cursul tratamentului cu alți inhibitori ai punctelor de control imunitar s-au raportat cazuri cu următoarele reacții adverse, care ar putea apărea și în cursul tratamentului cu tislelizumab: insuficiență pancreatică exocrină.
Reacții legate de perfuzare
La pacienții tratați cu tislelizumab în monoterapie, reacțiile legate de perfuzare au apărut la 3,0% dintre pacienți, inclusiv evenimente de grad 3 (0,1%). Tislelizumab a fost întrerupt definitiv la 0,1% dintre pacienți, iar tratamentul cu tislelizumab a fost întrerupt temporar la 0,1% dintre pacienți.
După punerea pe piață au fost raportate cazuri de anafilaxie, cu reacție anafilactică și șoc anafilactic.
Anomalii de laborator
La pacienții tratați cu tislelizumab în monoterapie, proporțiile pacienților care au prezentat modificări de la valorile inițiale la valori anormale de grad 3 sau 4 ale analizelor de laborator au fost după cum urmează: 0,1% pentru creșterea valorii hemoglobinei, 4,4% pentru scăderea valorii hemoglobinei, 0,9% pentru scăderea numărului de leucocite, 8,9% pentru scăderea numărului de limfocite, 0,2% pentru creșterea numărului de limfocite, 2,1% pentru scăderea numărului de neutrofile, 1,3% pentru scăderea numărului de trombocite, 2,6% pentru valori crescute ale alanin aminotransferazei, 0,3% pentru scăderea valorilor albuminei, 2,7% pentru valori crescute ale fosfatazei alcaline, 4,8% pentru creșterea aspartat aminotransferazei, 2,8% pentru creșterea valorii bilirubinei, 1,9% pentru creșterea valorii creatinkinazei, 1,2% pentru valori crescute ale creatininei, 4,4% pentru valori crescute ale glicemiei, 0,5% pentru valori crescute ale glicemiei, 0,9% pentru valori crescute ale potasiului, 2,9% pentru valori scăzute ale potasiului, 0,1% pentru valori crescute ale sodiului, 6,5% pentru valori scăzute ale sodiului.
La pacienții tratați cu tislelizumab în asociere cu chimioterapie, proporția de pacienți care au prezentat o schimbare de la valorile inițiale la valori anomale de grad 3 sau 4 a analizelor de laborator a fost după cum urmează: 14,2% pentru scăderea valorii hemoglobinei, 23,3% pentru scăderea numărului de leucocite, 17,9% pentru scăderea numărului de limfocite, 0,1% pentru creșterea numărului de limfocite, 47,2% pentru scăderea numărului de neutrofile, 14,1% pentru scăderea numărului de trombocite, 3,5% pentru valori crescute ale alanin aminotransferazei, 0,5% pentru scăderea valorilor albuminei, 0,8% pentru creșterea valorilor fosfatazei alcaline, 3,1% pentru creșterea valorilor aspartat aminotransferazei, 2,0% pentru valori crescute ale bilirubinei, 2,3% pentru valori crescute ale creatinkinazei, 1,8% pentru valori crescute ale creatininei, 0,5% pentru valori scăzute ale glucozei, 1,2% pentru valori crescute ale glucozei, 1,3% pentru valori crescute ale potasiului, 7,6% pentru valori scăzute ale potasiului, 0,3% pentru valori crescute ale sodiului și 11,5% pentru valori scăzute ale sodiului.
Imunogenitate
Dintre cei 3.614 pacienți evaluabili cu anticorpi antimedicament (antidrug antibodies - ADA), 21,1% dintre pacienți au fost testați pozitiv pentru ADA rezultați în urma tratamentului și au fost detectați anticorpi neutralizanți (NAbs) la 0,9% dintre pacienți. Analiza farmacocinetică la nivelul populației a arătat că statusul ADA a fost o covariată semnificativă din punct de vedere statistic în ceea ce privește clearance-ul; cu toate acestea, prezența ADA împotriva tislelizumab rezultați în urma tratamentului pare să nu aibă niciun impact semnificativ din punct de vedere clinic asupra farmacocineticii sau eficacității.
În rândul pacienților evaluabili cu anticorpi antimedicament cărora li s-au administrat 200 mg o dată la fiecare 3 săptămâni monoterapie sau în asociere cu chimioterapii (inclusiv adjuvant 400 mg o dată la 6 săptămâni în NSCLC rezecabil), au fost observate următoarele rate de apariție a evenimentelor adverse (adverse events – AEs) la populația cu anticorpi antimedicament comparativ cu populația fără anticorpi antimedicament, respectiv: evenimente adverse de grad ≥3, 52,5% comparativ cu 42,1%, 39,0% comparativ cu 31,8% evenimente adverse grave (serious adverse events – SAEs), 12,3% comparativ cu 11,4% (pentru monoterapie) evenimente adverse care au dus la întreruperea tratamentului cu tislelizumab; 80,0% comparativ cu 78,6% pentru EA de grad ≥ 3, 43,3% comparativ cu 41,0% pentru EAG, 13,6% comparativ cu 13,5% pentru EA care conduc la întreruperea tratamentului cu tislelizumab (pentru terapia în asociere).
La pacienții care au dezvoltat anticorpi antimedicament ca urmare a tratamentului s-a observat că aveau inițial o stare generală de sănătate mai precară și caracteristici ale bolii care pot conduce la confuzie în interpretarea analizei privind siguranța. Datele disponibile nu permit tragerea unor concluzii ferme cu privire la tipare posibile ale apariției reacțiilor adverse la medicament.
Vârstnici
La administrarea de tislelizumab în monoterapie sau în asociere cu chimioterapie nu au fost observate diferențe generale în ceea ce privește siguranța între pacienții cu vârsta <65 ani și pacienții cu vârsta cuprinsă între 65 și 74 ani. Datele pentru pacienții cu vârsta de 75 ani și peste această vârstă sunt prea limitate pentru a trage concluzii.
Raportarea reacțiilor adverse suspectate
Raportarea reacțiilor adverse suspectate după autorizarea medicamentului este importantă. Acest lucru permite monitorizarea continuă a raportului beneficiu/risc al medicamentului. Profesioniștii din domeniul sănătății sunt rugați să raporteze orice reacție adversă suspectată prin intermediul sistemului național de raportare, astfel cum este menționat în Anexa V.
4.9 Supradozaj
Nu există informații despre supradozajul cu tislelizumab. În caz de supradozaj, pacienții trebuie monitorizați atent pentru semne sau simptome ale reacțiilor adverse la medicament și trebuie instituit imediat un tratament simptomatic adecvat.
5. PROPRIETĂȚI FARMACOLOGICE
5.1 Proprietăți farmacodinamice
Grupa farmacoterapeutică: Agenți antineoplazici, anticorpi monoclonali și conjugați anticorpmedicament, codul ATC: L01FF09
Mecanism de acțiune
Tislelizumab este o variantă de anticorp monoclonal umanizat împotriva PD-1de tip imunoglobulină G4 (IgG4), care se leagă de domeniul extracelular al PD-1 uman. Acesta blochează competitiv legarea atât a PD-L1, cât și a PD-L2, inhibând semnalizarea negativă mediată de PD-1 și îmbunătățind activitatea funcțională la nivelul celulelor T în analizele celulare in vitro.
Eficacitate și siguranță clinică
Pe baza modelului și a simulării relațiilor dintre expunere și răspuns pentru eficacitatea și siguranța tislelizumabului, nu există diferențe semnificative clinic de eficacitate sau siguranță între dozele de 200 mg o dată la 3 săptămâni și de 400 mg o dată la 6 săptămâni.
Cancer pulmonar altul decât cel cu celule mici (NSCLC)
Tratamentul neoadjuvant și adjuvant al NSCLC rezecabil: BGB-A317-315
BGB-A317-315 a fost un studiu de fază 3, randomizat, controlat cu placebo, în regim dublu-orb, pentru investigarea eficacității și a siguranței tratamentului neoadjuvant cu tislelizumab în asociere cu chimioterapie dublă pe bază de platină urmat de tratament adjuvant cu tislelizumab comparativ cu
tratamentul neoadjuvant cu placebo plus chimioterapie dublă pe bază de platină urmat de tratament adjuvant cu placebbo la pacienți cu NSCLC rezecabil în stadiul II sau IIIA.
Studiul a inclus pacienți cu NSCLC în stadiul II sau IIIA (conform clasificării AJCC [American Joint Committee on Cancer], ediția 8) confirmat histologic, cu status de performanță ECOG de 0 sau 1 și fără mutații EGFR sau translocații ale genei ALK cunoscute și eligibilitate confirmată pentru rezecție R0 cu intenție curativă. Nu au fost incluși în studiu pacienți cu NSCLC în stadiul IIIB.
Următoarele criterii de selecție definesc pacienții cu risc crescut de recurență care sunt incluși în indicația terapeutică și reflectă populația de pacienți cu stadiul II – IIIA conform sistemului de stadializare AJCC, ediția a 8a:
- Dimensiune a tumorii > 4 cm; sau tumori de orice dimensiune însoțite de status N1 sau N2;
- Tumori care invadează structurile toracice (invadează direct pleura viscerală, pleura parietală, peretele toracic, bronhiile principale, nervul frenic, pleura mediastinală, pericardul parietal);
- Tumori > 4 cm care cauzează atelectazie obstructivă ce se extinde în regiunea hilară, care afectează o parte din plămân sau întreg plămânul sau care afectează o bronhie principală, indiferent de distanța față de carină, sau care invadează pleura viscerală (PL1 sau PL2) pentru statusul N0;
- Tumori cu nodul(i) separat(ți) în același lob ca cancer pulmonar primar.
În total 453 de pacienți au fost randomizați (1:1) pentru a li se administra
- Brațul cu tislelizumab: tislelizumab neoadjuvant 200 mg în Ziua 1 în asociere fie cu cisplatină 75 mg/m2 fie cu carboplatină ASC 5 mg/ml/minut și pemetrexed 500 mg/m2 sau paclitaxel 175 mg/m2 în Ziua 1 a fiecărui ciclu de 21 de zile, timp de 3 sau 4 cicluri. După intervenția chirurgicală, s-a administrat tislelizumab adjuvant 400 mg o dată la 6 săptămâni timp de până la 8 cicluri.
- Brațul cu placebo: placebo neoadjuvant în Ziua 1 în asociere fie cu cisplatină 75 mg/m2 fie cu carboplatină ASC 5 mg/ml/minut și pemetrexed 500 mg/m2 sau paclitaxel 175 mg/m2 în Ziua 1 a fiecărui ciclu de 21 de zile, timp de 3 sau 4 cicluri. După intervenția chirurgicală, s-a administrat placebo adjuvant o dată la 6 săptămâni timp de până la 8 cicluri.
Pacienților cu histologie non-scuamoasă li s-a administrat pemetrexed, în timp ce pacienților cu histologie scuamoasă li s-a administrat paclitaxel, în timp ce alegerea cisplatinei sau carboplatinei a fost decisă de investigatori pentru toți pacienții. Dacă a fost indicată, pacienților li s-a administrat radioterapie adjuvantă postoperatorie înainte de tratamentul adjuvant cu tislelizumab sau placebo. Administrarea tislelizumabului și a chimioterapiei a continuat până la finalizarea tratamentului, progresia bolii, apariția EA inacceptabile, deces, sau decizia pacientului și/sau a investigatorului de a întrerupe tratamentul de studiu.
Criteriile finale de evaluare primare duale au fost supraviețuirea fără evenimente (SFE) conform unei evaluări independente efectuate la nivel central în regim orb (blinded independent central review – BICR) și rata de răspuns patologic major (RPM) conform unei evaluări patologice independente în regim orb (blinded independent pathological review – BIPR). Criteriile finale de evaluare secundare privind eficacitatea au inclus rata de răspuns complet patologic (RCp) conform BIPR și supraviețuirea generală (SG).
Datele demografice și caracteristicile inițiale au fost în general echilibrate între cele 2 brațe de tratament. Caracteristicile inițiale pentru toți cei 453 de pacienți randomizați au fost: vârsta mediană de 62 de ani (interval: 30 până la 80 de ani); 40% dintre pacienți aveau vârsta ≥65 de ani; 3,3% dintre pacienți aveau vârsta ≥75 de ani; 90,5% dintre pacienți erau bărbați; 100% asiatici (toți înrolați în China), 65,3% aveau un scor de performanță ECOG de 0; 84,5% erau fumători actuali sau foști fumători; 78,1% aveau histologie scuamoasă diagnosticată; 58,5% aveau boală în stadiul IIIA; 57,8% aveau o expresie a PD-L1 ≥1%.
84,1% dintre pacienții din brațul cu tislelizumab în asociere cu chimioterapie care conține săruri de platină au suportat o intervenție chirurgicală definitivă, comparativ cu 76,2% dintre pacienții din brațul cu chimioterapie care conține săruri de platină.
Studiul a demonstrat o îmbunătățire semnificativă statistic a RPM, SFE, RCp și SG la pacienții randomizați în brațul cu tislelizumab comparativ cu brațul cu placebo.
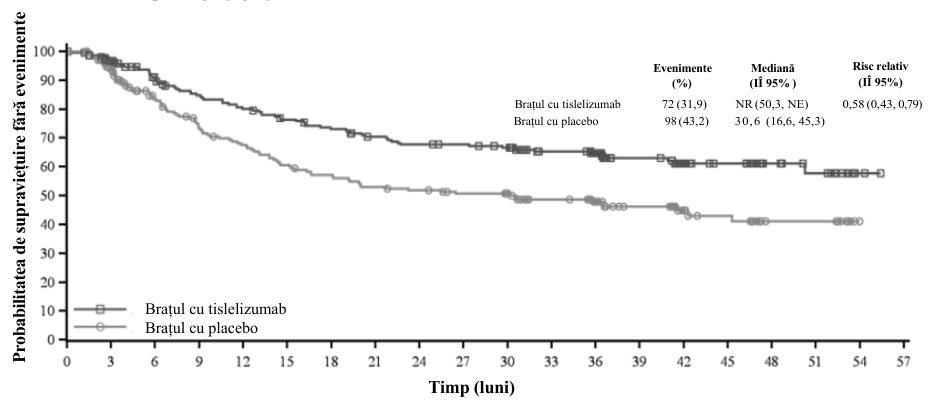
La analiza interimară prespecificată a SFE (data centralizării datelor: 21 august 2023), RR pentru SFE a fost de 0,56 (IÎ 95%: 0,40, 0,79, valoare p unilaterală de 0,0003), iar perioada mediană de urmărire a SG conform metodologiei Kaplan-Meier inverse a fost de 24,6 luni în brațul cu tislelizumab și 22,7 luni în brațul cu placebo.
Tabelul 3, Figura 1 și Figura 2 sintetizează rezultatele privind eficacitatea.
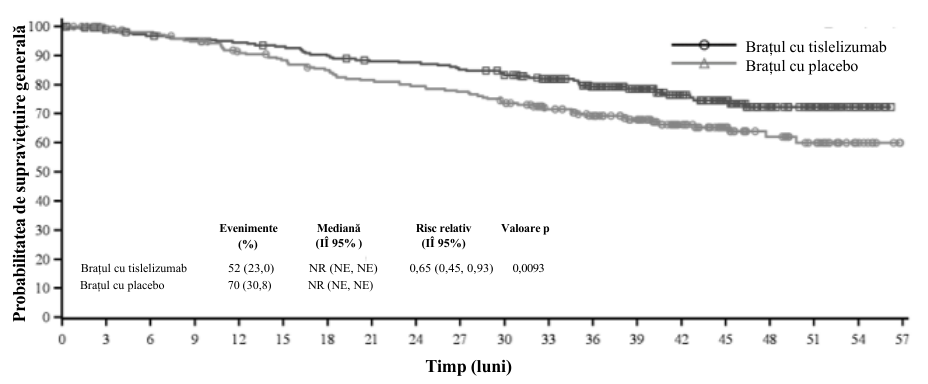
La analiza finală prespecificată (data centralizării datelor: 07 martie 2025), perioada mediană de urmărire a SG conform metodologiei Kaplan-Meier inverse a fost de 43,3 luni (IÎ 95%: 41,2, 44,6) în brațul cu tislelizumab și 41,6 luni (IÎ 95%: 39,9, 43,8) în brațul cu placebo.
Tabelul 3 Rezultate privind eficacitatea în cadrul studiului BGB-A317-3151
| Brațul cu tislelizumab (N = 226) | Brațul cu placebo (N = 227) | ||
| Supraviețuire fără evenimente | |||
|---|---|---|---|
| Evenimente, n (%) | 72 (31,9) | 98 (43,2) | |
| Mediană (luni) (IÎ 95%) | NR (50,3, NE) | 30,6 (16,6, 45,3) | |
| RR (IÎ 95%)a | 0,58 (0,43, 0,79) | ||
| Răspuns patologic major | |||
| n (%) | 127 *56,2) | 34 (15) | |
| IÎ 95%c | (49,5, 62,8) | (10,6, 20,3) | |
| Diferență, % (IÎ 95%)d | 41,1 (33,2, 49,1) | ||
| Valoare pe | <0,0001 | ||
| Supraviețuire generală | |||
| Decese, n (%) | 52 (23,0) | 70 (30,8) | |
| Mediană (luni) (IÎ 95%) | NR (NE, NE) | NR (NE, NE) | |
| RR (IÎ 95%)a | 0,65 (0,45, 0,93) | ||
| Valoare pe | 0,0093 | ||
IÎ = interval de încredere; RR = risc relativ, NE = nu se poate estima, NR = nu a fost atinsă
Pacienții fără intervenție chirurgicală sau rezultate patologice au fost considerați nerespondenți.
1 Analiza finală prespecificată a RPM s-a bazat pe datele colectate până la data limită de 20 februarie 2023, iar analiza finală prespecificată a SFE și a SG s-a bazat pe datele colectate până la data limită de 07 martie 2025. a Riscul relativ și IÎ de 05% au fost estimate utilizând un model de regresie Cox stratificat în funcție de histologie, stadiul bolii și expresia PD-L1 din tehnologia de răspuns interactiv (IRT). b Valoare p a fost calculată utilizând un test de tip log-rank stratificat în funcție de histologie, stadiul bolii și expresia PD-L1 din IRT. c IÎ 95% a fost estimat utilizând metoda Clopper-Pearson. d Diferența în de risc comun Mantel-Haenszel a fost estimată împreună cu IÎ de 95% aferente construite printr-o aproximare normală și estimatorul de varianță Sato stratificat în funcție de histologie, stadiul bolii și expresia PD-L1 din IRT. e Valoarea p a fost obținută prin metoda Cochral-Mantel-Haenszel stratificată în funcție de histologie, stadiul bolii și expresia PD-L1 din IRT.
Figura 1 Grafic Kaplan-Meier pentru supraviețuirea fără evenimente în studiul
BGB-A317-315
Număr la risc
Brațul cu tislelizum 226 196 176 161 152 143 136 128 123 121 117 101 92 69 49 39 21 17 2 0 ab
Brațul cu
227 187 149 128 117 105 98 91 88 83 79 69 59 47 29 22 11 11 0 0 placebo
Figura 2 Grafic Kaplan-Meier pentru supraviețuirea generală în studiul BGB-A317-315
Număr la risc
Brațul cu tislelizum 226 218 212 209 206 202 195 189 188 183 176 163 143 121 91 69 47 36 15 0 ab
Brațul cu
227 214 207 199 186 180 172 165 161 157 148 131 117 98 73 51 34 26 9 0 placebo
O analiză pe subgrupuri a fost efectuată în studiul BGB-A317-315 la pacienții cu PD-L1 ≥1% (brațul cu tislelizumab [n=130; 58%] față de brațul cu placebo [n=132; 58%]) și PD-L1 <1% (care exclude o valoare neevaluabilă/nedeterminată) (brațul cu tislelizumab [n=89; 39%] față de brațul cu placebo [n=84; 37%]). RR pentru SFE a fost de 0,53 (IÎ 95%: 0,35, 0,79) la pacienții cu PD-L1 ≥1% și 0,70 (IÎ 95%: 0,43, 1,14) la pacienții cu PD-L1 < 1%. RR pentru SG a fost de 0,61 (IÎ 95%: 0,38, 0,98) la pacienții cu PD-L1 ≥1% și 0,91 (IÎ 95%: 0,50, 1,64) la pacienții cu PD-L1 <1%.
Tratamentul de primă intenție al NSCLC non-scuamos: BGB-A317-304
BGB-A317-304 a fost un studiu multicentric, randomizat, deschis, de fază 3, care a investigat eficacitatea și siguranța tislelizumab în asociere cu platină-pemetrexed comparativ cu platină-pemetrexed în monoterapie ca tratament de primă intenție la pacienții cărora nu li s-a administrat anterior chimioterapie, cu NSCLC non-scuamos în stadiu avansat local, care nu au fost eligibili pentru rezecție chirurgicală sau chimioterapie pe bază de platină, sau cu NSCLC non-scuamos în stadiu metastatic.
Studiul a exclus pacienții cu metastaze cerebrale sau leptomeningiene active, mutații EGFR cunoscute sau translocații ALK sensibile la terapiile țintite cu inhibitori disponibile, boală autoimună activă sau orice afecțiune care necesită tratament sistemic fie cu corticosteroizi (>10 mg de prednison zilnic sau echivalent), fie cu alte imunosupresoare.
Un total de 334 pacienți au fost randomizați (2:1) pentru a li se administra tislelizumab 200 mg în asociere cu pemetrexed 500 mg/m2 și carboplatin ASC 5 mg/ml/minut sau cisplatin 75 mg/m2 (brațul T+PP, N = 223) sau pemetrexed 500 mg/m2 și carboplatin ASC 5 mg/ml/minut sau cisplatin 75 mg/m2 (brațul PP, N = 111). Alegerea platinei (cisplatin sau carboplatin) a fost decizia investigatorului.
Tratamentul a fost administrat în decursul unui ciclu de 3 săptămâni. După administrarea a 4, 5 sau 6 cicluri de chimioterapie sau tislelizumab în asociere cu chimioterapie, la decizia investigatorului, pacienților din brațul T+PP li s-a administrat tislelizumab 200 mg în asociere cu pemetrexed 500 mg/m2 în decursul unui ciclu de 3 săptămâni, până la progresia bolii sau apariția toxicității inacceptabile; pacienților din brațul PP li s-a administrat pemetrexed 500 mg/m2 în monoterapie până la progresia bolii sau apariția toxicității inacceptabile, iar celor cu progresie a bolii confirmată de Comitetul Independent de Examinare (Independent Review Committee - IRC) li s-a oferit opțiunea de a trece la administrarea tislelizumab în monoterapie în decursul unui ciclu de 3 săptămâni.
Randomizarea a fost stratificată în funcție de expresia PD-L1 în celulele tumorale (tumour cells - TC) (<1% față de 1% până la 49% față de ≥50%) și stadiul bolii (IIIB față de IV), conform clasificării din American Joint Committee on Cancer (AJCC), ediția a 7-a a Cancer Staging Manual. Expresia PD-L1 a fost evaluată într-un laborator central, folosind testul Ventana PD-L1 (SP263), care a identificat, printr-o tehnică de colorare, prezența PD-L1 la nivelul celulelor tumorale. Evaluările au fost efectuate la fiecare 6 săptămâni în primele 6 luni, apoi la fiecare 9 săptămâni în următoarele 6 luni, apoi la fiecare 12 săptămâni.
Caracteristicile de bază ale pacienților din studiul BGB-A317-304 au fost: vârsta mediană de 61 de ani (interval: 25-75), 29% vârsta de 65 ani sau peste această vârstă; 74% bărbați; 100% asiatici (toți înrolați în China); 23,4% cu ECOG PS de 0 și 76,6% cu ECOG PS de 1; 18,3% cu stadiu de boală IIIB; 26,6% cu status necunoscut al rearanjării ALK și 73,4% cu rearanjare ALK negativă; 36,2% nu au fumat niciodată; 5,4% cu metastaze cerebrale. Caracteristicile de vârstă, sex, status ECOG PS, stadializare, fumător, scor TC PD-L1 și tratamente antineoplazice anterioare au fost distribuite uniform între brațele de tratament.
Obiectivul final principal privind eficacitatea a fost supraviețuirea fără progresia bolii (SFP) conform RECIST v1.1 și conform IRC în cadrul analizei intenției de tratament (intent to treat - ITT). Obiectivele secundare privind eficacitatea au inclus supraviețuirea globală (SG), rata de răspuns obiectiv (RRO) și durata răspunsului (DR) conform IRC și conform investigatorului.
Studiul și-a atins obiectivul final principal la analiza interimară (data centralizării datelor de 23 ianuarie 2020), prin ameliorarea semnificativă statistic a SFP la nivelul T+PP în comparație cu PP. Riscul relativ stratificat a fost de 0,65 (IÎ 95%: 0,47, 0,91; p = 0,0054), cu o medie a SFP de 9,7 luni cu T+PP și 7,6 luni cu PP. Perioadele mediane de urmărire a SG pe baza metodologiei Kaplan-Meier inverse au fost de 9,9 luni în brațul cu T+PP și de 9,7 luni în brațul cu PP.
Rezultatele eficacității din analiza finală (data centralizării datelor de 26 octombrie 2020) au coincis cu cele ale analizei interimare. La analiza finală, perioadele mediane de urmărire a SG pe baza metodologiei Kaplan-Meier inverse au fost de 18,4 luni în brațul cu T+PP și de 18,0 luni în brațul cu PP.
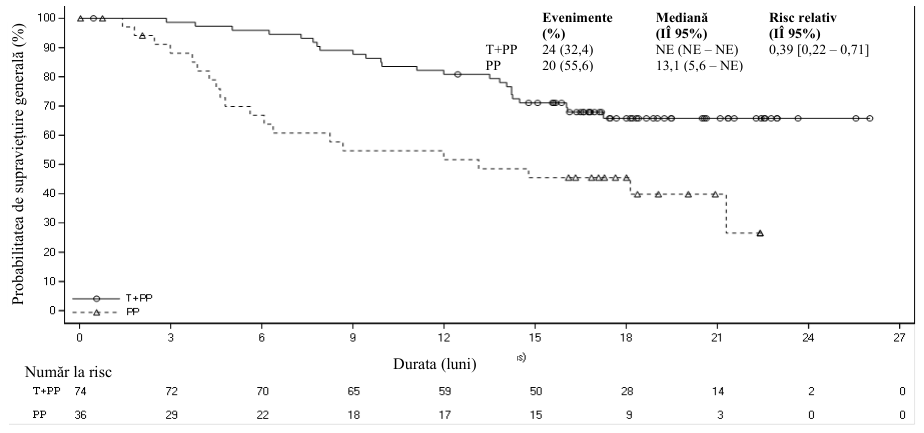
Dintre cei 334 de pacienți din studiul BGB-A317-304, 110 pacienți, respectiv (33%), au avut o expresie a PD-L1 în celulele tumorale ≥50%. Dintre aceștia, 74 de pacienți s-au aflat în grupul cu tislelizumab și chimioterapie, iar 36 de pacienți s-au aflat în grupul cu placebo și chimioterapie. Rezultatele eficacității la pacienții cu expresie a PD-L1 în celulele tumorale ≥50% din analiza finală sunt prezentate în Tabelul 4 și curba Kaplan-Meier pentru SFP și SG sunt prezentate în Figurile 3 și, respectiv, 4.
Tabelul 4 Rezultate privind eficacitatea în cadrul studiului BGB-A317-304 la pacienți cu expresie a PD-L1 ≥50%
| Obiectiv final | Tislelizumab + Pemetrexed + Platină (n = 74) | Pemetrexed + Platină (n = 36) |
|---|---|---|
| SFP | ||
| Evenimente, n (%) | 33 (44,6) | 22 (61,1) |
| SFP mediană (luni) (IÎ 95%) | 14,6 (11,5, NE) | 4,6 (3,5, 9,7) |
| Risc relativ stratificata (IÎ 95%) | 0,31 (0,18, 0,55) | |
| SG | ||
| Decese, n (%) | 24 (32,4) | 20 (55,6) |
| SG mediană (luni) (IÎ 95%) | NE (NE, NE) | 13,1 (5,6, NE) |
| Risc relativ stratificata (IÎ 95%) | 0,39 (0,22, 0,71) | |
| Cel mai bun răspuns general, n (%)b | ||
| RROb, n (%) | 52 (70,3) | 11 (30,6) |
| IÎ 95%c | (58,5, 80,3) | (16,3, 48,1) |
| DRb | ||
| DR mediană (luni) (IÎ 95%) | NE (13,2, NE) | 8,5 (3,3, NE) |
SFP = supraviețuire fără progresia bolii; IÎ = interval de încredere; SG = supraviețuirea generală; RRO = rata răspunsului obiectiv; DR = durata răspunsului; NE = nu se poate estima. Medianele au fost estimate prin metoda Kaplan-Meier, cu IÎ 95% estimat utilizând metoda Brookmeyer și Crowley. a Riscul relativ a fost estimat din modelul stratificat Cox cu grupa în care s-a administrat pemetrexed+platină ca grupă de referință și stratificat în funcție de stadiul bolii (IIIB față de IV). b SFP s-a bazat pe evaluarea IRC și RRO/DR s-a bazat pe răspunsul confirmat de IRC. c IÎ 95% a fost calculat utilizând metoda Clopper-Pearson. | ||
Figura 3 Grafic Kaplan-Meier pentru SFP în BGB-A317-304 la pacienți cu PD-L1 ≥50%
| Evenimente | Mediană Events Median Risc relativ Hazard ratio (%) (95% CI) (95% CI) |
|---|---|
| (%) | (IÎ 95%) T+PP 33(44.6) 14.6(11.5-NE) (IÎ 95%) 0.313[0.178- 0.547] |
| T+PP 33 (44,6) | 14,6 (11,5 PP – NE) 22(61.1) 4.6(3.5-9.7) 0,31 [0,18 – 0,55] |
| PP 22 (61,1) | 4,6 (3,5 – 9,7) |
| T+PP T+PP | |
| PP PP | |
100 100
Probabilitatea de surpraviețuire fără progresia bolii (%)
90 90
80 80
Progression-Free Survival Probability(%) Progression-Free Survival Probability(%)
70 70
60 60
50 50
40 40
30 30
20 20
10 10
0 0
0 0 3 3 6 6 9 9 12 12 15 15 18 18 21 21 24 24 27 27 Durata (luni) Time (M onths) Time (M onths)
Număr la risc
No. at Risk
T+PP 74 64 53 45 36 21 9 3 2 0 PP 36 18 9 7 4 1 1 0 0 0
Figura 4 Grafic Kaplan-Meier pentru SG în BGB-A317-304 la pacienții cu PD-L1 ≥50%
Tratamentul de primă intenție al NSCLC non-scuamos: BGB-A317-307
BGB-A317-307 a fost un studiu multicentric, randomizat, deschis, de fază 3, desfășurat pentru a compara eficacitatea și siguranța tislelizumab în asociere cu paclitaxel plus carboplatin sau nab-paclitaxel plus carboplatin comparativ cu paclitaxel plus carboplatin în monoterapie, ca tratament de primă intenție la pacienții cărora nu li s-a administrat anterior chimioterapie, cu NSCLC nonscuamos în stadiu avansat local, care nu au fost eligibili pentru rezecție chirurgicală sau chimioterapie pe bază de platină, sau cu NSCLC non-scuamos în stadiu metastatic.
Studiul a exclus pacienții cu metastaze cerebrale sau leptomeningiene active, mutații EGFR cunoscute sau translocații ALK sensibile la terapiile țintite cu inhibitori disponibile, boală autoimună activă sau orice afecțiune care necesită tratament sistemic fie cu corticosteroizi (>10 mg de prednison zilnic sau echivalent), fie cu alte imunosupresoare.
Un total de 360 pacienți au fost randomizați (1:1:1) pentru a li se administra tislelizumab 200 mg în asociere cu paclitaxel 175 mg/m2 și carboplatin ASC 5 mg/ml/min (brațul de tratament T+PC, N = 120), sau tislelizumab 200 mg în asociere cu nab-paclitaxel 100 mg/m2 și carboplatin ASC 5 mg/ml/minut (brațul de tratament T+nPC, N = 119), sau paclitaxel 175 mg/m2 și carboplatin ASC 5 mg/ml/minut (brațul de tratament PC, N = 121).
Tratamentul a fost administrat într-un ciclu de 3 săptămâni, până când pacientul a finalizat administrarea a 4 până la 6 cicluri de chimioterapie sau tislelizumab în asociere cu chimioterapie, la decizia investigatorului. Pacienților din brațele de tratament T+nPC și T+PC li s-a administrat tislelizumab până la progresia bolii sau apariția toxicității inacceptabile. Pacienților din brațul PC care au prezentat progresia bolii li s-a oferit opțiunea de a trece la administrarea tislelizumab în monoterapie într-un ciclu de 3 săptămâni.
Randomizarea a fost stratificată în funcție de expresia PD-L1 în celulele tumorale (TC) (<1% față de 1% până la 49% față de ≥50%) și stadiul bolii (IIIB față de IV), conform clasificării din American Joint Committee on Cancer (AJCC), ediția a 7a a Cancer Staging Manual. Expresia PD-L1 a fost evaluată într-un laborator central, folosind testul Ventana PD-L1 (SP263), care a identificat, printr-o tehnică de colorare, prezența PD-L1 la nivelul celulelor tumorale. Evaluările au fost efectuate la fiecare 6 săptămâni pentru primele 6 luni, apoi la fiecare 9 săptămâni pentru restul perioadei din primul an, apoi la fiecare 12 săptămâni până la progresia bolii.
Caracteristicile de bază ale populației în studiu au fost: vârsta mediană de 62.0 ani (interval: 34 până la 74), 35,3% vârsta de 65 ani sau peste această vârstă; 91,7% bărbați; 100% asiatici (toți înrolați în China), 23,6% cu ECOG PS de 0 și 76,4% cu ECOG PS de 1; 33,9% diagnosticați în stadiul IIIB și 66,1% în stadiul IV la momentul inițial; 16,4% nu au fumat niciodată; 38,3% cu scor TC PD-L1 <1%, 25,3% cu scor TC PD-L1 ≥1% și ≤49%, 34,7% cu scor TC PD-L1 ≥50%. Caracteristicile de vârstă, sex, status ECOG PS, stadializare, fumător, scor TC PD-L1 și tratamente antineoplazice anterioare au fost distribuite uniform între brațele de tratament.
Obiectivul final principal privind eficacitatea a fost supraviețuirea fără progresia bolii (SFP) conform evaluării IRC conform RECIST v1.1 în analiza ITT, care a fost testată secvențial în brațele T+PC față de PC și brațele T+nPC față de PC. Obiectivele secundare privind eficacitatea au inclus supraviețuirea globală (SG), rata de răspuns obiectiv (RRO) și durata răspunsului (DR) conform IRC și conform investigatorului.
Studiul și-a atins obiectivul final principal la analiza interimară (data centralizării datelor de 6 decembrie 2019), prin ameliorarea semnificativă statistic a SFP la administrarea tislelizumab în asociere cu paclitaxel și carboplatin (brațul T+PC) și a tislelizumab în asociere cu nab-paclitaxel și carboplatin (brațul T+nPC) în comparație cu paclitaxel și carboplatin în monoterapie (brațul PC). RR stratificat (brațul T+PC față de brațul PC) a fost de 0,48 (95% ÎI: 0,34, 0,69; p <0,0001). RR stratificat (brațul T+nPC față de brațul PC) a fost de 0,45 (95% CI: 0,32, 0,64; p <0,0001). SFP mediană a fost de 7,6 luni în brațul T+PC, 7,6 luni în brațul T+nPC și 5,4 luni în brațul PC. Perioadele mediane de urmărire a SG pe baza metodologiei Kaplan-Meier inverse au fost de 8,8 luni în brațul cu T+PC, de 8,8 luni în brațul cu T+nPC și de 8 luni în brațul cu PC.
Analiza finală (data centralizării datelor de 30 septembrie 2020) a demonstrat rezultatele constante, comparabile cu cele ale analizei interimare. La analiza finală, perioadele mediane de urmărire a SG pe baza metodologiei Kaplan-Meier inverse au fost de 18,8 luni în brațul cu T+PC, de 18,9 luni în brațul cu T+nPC și de 18,1 luni în brațul cu PC.
Rezultatele privind eficacitatea la nivelul analizei finale sunt indicate în Tabelul 5, Figura 5 și Figura 6.
Tabelul 5 Rezultate privind eficacitatea în cadrul studiului BGB-A317-307
| Obiectiv final | Tislelizumab + Paclitaxel + Carboplatin (N = 120) | Tislelizumab + nab-Paclitaxel + Carboplatin (N = 119) | Paclitaxel + Carboplatin (N = 121) |
|---|---|---|---|
| SFP | |||
| Evenimente, n (%) | 80 (66,7) | 79 (66,4) | 86 (71,1) |
| SFP mediană (luni) (IÎ 95%) | 7,7 (6,7, 10,4) | 9,6 (7,4, 10,8) | 5,5 (4,2, 5,6) |
| Risc relativ stratificata (IÎ 95%) | 0,45 (0,33, 0,62) | 0,43 (0,31, 0,60) | - |
| SG | |||
| Decese, n (%) | 48 (40,0) | 47 (39,5) | 52 (43,0) |
| SG mediană (luni) (IÎ 95%) | 22,8 (19,1, NE) | NE (18,6, NE) | 20,2 (16,0, NE) |
| Risc relativ stratificat (IÎ 95%) | 0,68 (0,45, 1,01) | 0,752 (0,50, 1,12) | - |
| RROb | |||
| RRO, n (%) | 74 (61,7) | 74 (62,2) | 45 (37,2) |
| IÎ 95% | (52,4, 70,4) | (52,8, 70,9) | (28,6, 46,4) |
| DRb | |||
| DR mediană (luni) (IÎ 95%) | 13,2 (7,85, 18,79) | 10,4 (8,34, 17,15) | 4,8 (4,04, 5,72) |
| SFP = supraviețuire fără progresia bolii; IÎ = interval de încredere; SG = supraviețuirea generală; RRO = rata răspunsului obiectiv; DR = durata răspunsului; NE = nu se poate estima. a Stratificat conform factorilor de stratificare: stadiul bolii (IIIB față de IV) și expresia PD-L1 în celula tumorală (≥50% TC față de 1% până la 49% TC față de <1% TC). b SFP s-a bazat pe evaluarea IRC, iar RRO/DR s-a bazat pe răspunsul confirmat de IRC | |||
Figura 5 Grafic Kaplan-Meier pentru SFP în cadrul studiului BGB-A317-307 conform IRC
Brațul T+PC față de brațul T+nPC față de brațul PC
100 90 T+PC: Evenimente (%) = 80 (66,7) Mediană (IÎ 95%): 7,7 (6,7-10,4) T+PC: Events(%) = 80(66.7) M edian(95% CI): 7.7(6.7-10.4) T+nPC: Evenimente (%) = 79 (66,4) Mediană (IÎ 95%): 9,6 (7,4-10,8) T+nPC: Events(%) = 79(66.4) M edian(95% CI): 9.6(7.4-10.8) Progression-Free Survival Probability(%) 80 PC: Evenimente (%) = 86 (71,1) Mediană (IÎ 95%): 5,5 (4,2-5,6) PC: Events(%) = 86(71.1) M edian(95% CI): 5.5(4.2-5.6) 70 progresia bolii (%) RR (IÎ 95%) T+PC vs. PC = 0,45 [0,33-0,62] HR(95 % CI)T+PC vs. PC= 0.450 [0.326- 0.619] 60 RR (IÎ 95%) T+nPC vs. PC = 0,43 [0,31-0,6] HR(95 % CI)T+nPC vs. PC= 0.428 [0.308- 0.595] 50 40 30 20 10 0 0 3 6 9 12 15 18 21 24 Timp (luni) Time (M onths) Număr de pacienți care încă prezintă risc Number of patients still at risk Timp Time 0 3 6 9 12 15 18 21 24 T+PC T+PC 120 97 66 51 37 27 13 2 0 T+nPC T+nPC 119 99 66 55 31 20 15 3 0 PC PC 121 74 31 13 8 5 4 1 0 | |
| Probabilitatea supraviețuirii fără | Progression-Free Survival Probability(%) progresia bolii (%) |
| Timp Time T+PC T+PC T+nPC T+nPC PC PC |
IÎ = interval de încredere; T+PC = tislelizumab+paclitaxel+carboplatin; T+nPC = tislelizumab+nab-paclitaxel+carboplatin; PC = paclitaxel+carboplatin.
Figura 6 Grafic Kaplan-Meier pentru SFP în cadrul studiului BGB-A317-307
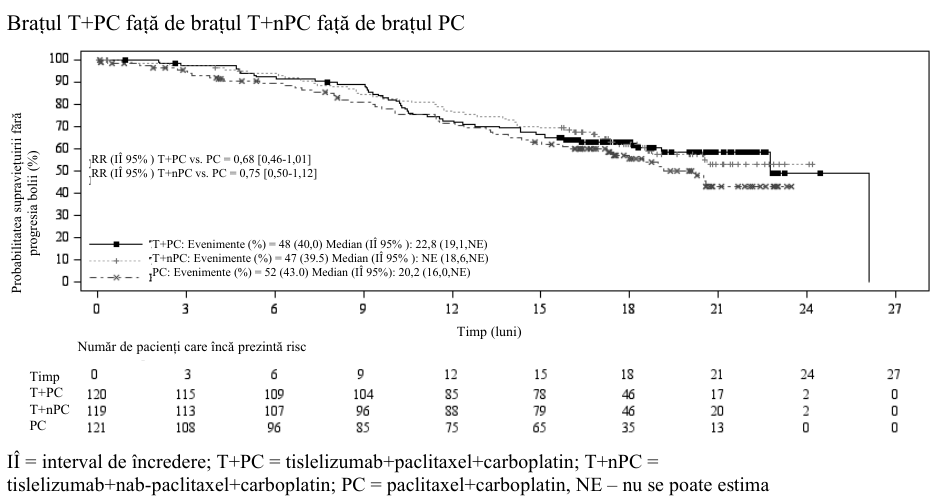
Analizele de subgrup au demonstrat un efect constant al tratamentului în ce privește SFP în subgrupurile demografice și de prognostic majore, inclusiv expresia PD-L1 <1%, 1 până la 49% și ≥50% și stadiile bolii IIIB și IV:
- pentru T+PC, cu SFP a SG de 0,57 (95% ÎI, RR = 0,34, 0,94) pentru PD-L1 <1%, 0,40 (95% ÎI, RR = 0,21, 0,76) pentru 1 până la 49% și 0,44 (95% ÎI, RR = 0,26, 0,75) pentru ≥50%
- pentru T+nPC, cu SPF a SG de 0,65 (95% ÎI, RR = 0,40, 1,06) pentru PD-L1 <1%, 0,40 (95% ÎI, RR = 0,22, 0,74) pentru 1 până la 49% și 0,33 (95% CI, RR = 0,18, 0,59) pentru ≥50%
NSCLC tratat anterior: BGB-A317-303
BGB-A317-303 a fost un studiu multicentric, randomizat, deschis, de fază 3, care a investigat eficacitatea și siguranța tislelizumab comparativ cu docetaxel la pacienții cu NSCLC în stadiu avansat local (scuamos sau non-scuamos), care au prezentat progresia bolii pe parcursul administrării unei scheme anterioare de tratament pe bază de platină sau după administrarea acesteia.
Studiul a exclus pacienții cu mutații EGFR cunoscute sau translocații ALK, cărora li s-a administrat anterior tratament cu inhibitori ai PD-(L)1 sau tratament cu inhibitori ai CTLA-4, boală autoimună activă sau orice afecțiune care necesită tratament sistemic fie cu corticosteroizi (>10 mg de prednison zilnic sau echivalent), fie cu alte imunosupresoare.
În total 805 pacienți au fost randomizați (2:1) pentru a li se administra tislelizumab 200 mg intravenos, la interval de 3 săptămâni (n = 535) sau docetaxel 75 mg/m2 intravenos, la interval de 3 săptămâni (n = 270). Randomizarea a fost stratificată în funcție de histologie (scuamos față de non-scuamos), linii terapeutice (de a doua intenție sau de a treia intenție) și expresia PD-L1 la nivelul celulelor tumorale (TC) (≥25% față de <25%). Administrarea docetaxel și tislelizumab a continuat până la progresia bolii, conform evaluării de către investigator și conform RECIST v1.1 sau până la apariția toxicității inacceptabile. Expresia PD-L1 a fost evaluată într-un laborator central, folosind testul Ventana PD-L1 (SP263), care a identificat, printr-o tehnică de colorare, prezența PD-L1 la nivelul celulelor tumorale. Evaluările tumorale au fost efectuate la fiecare 9 săptămâni timp de 52 săptămâni după randomizare, și au continuat ulterior la interval de 12 săptămâni. Statusul supraviețuirii a fost urmărit la fiecare 3 luni de la întreruperea tratamentului de studiu.
Caracteristicile de bază ale populației în studiu au fost: vârsta mediană de 61 ani (interval: 28 până la 88), 32,4% vârsta de 65 ani sau peste această vârstă, 3,2% vârsta de 75 ani sau peste această vârstă; 77,3% bărbați; 17,0% caucazieni și 79,9% asiatici; 20,6% cu status ECOG PS de 0 și 79,4% cu status ECOG PS de 1; 85,5% cu boală în stadiu metastatic; 30,3% nu au fumat niciodată; 46,0% cu histologie scuamoasă și 54,0% cu histologie non-scuamoasă; 65,8% cu status EGFR de tip sălbatic și 34% cu status EGFR necunoscut; 46,1% cu status ALK de tip sălbatic și 53,9% cu status ALK necunoscut; 7,1% cu metastaze cerebrale tratate anterior.
57,0% dintre pacienți au prezentat un scor PD-L1 TC <25% și 42,5% au prezentat un scor PD-L1 TC ≥25%. Tuturor pacienților li s-a administrat anterior o schemă dublă de tratament cu platină: la 84,7% dintre pacienți s-a administrat anterior o schemă de tratament, la 15,3% s-au administrat anterior două scheme de tratament.
Obiectivele finale principale duble privind eficacitatea au fost SG în seturile de analiză ITT și scorul TC PD-L1 ≥25%. Obiectivele finale suplimentare privind eficacitatea au inclus SFP, RRO și DR evaluate de investigator.
BGB-A317-303 a îndeplinit obiectivele finale principale duble ale SG în seturile de analiză ITT și PD-L1 ≥25%. La analiza interimară prespecificată (data centralizării datelor de 10 august 2020), populația ITT a prezentat ameliorarea semnificativă statistic a SG. Rezultatele au favorizat brațul cu tislelizumab (stratificare RR = 0,64; IÎ 95%: 0,53, 0,78; p < 0,0001). SG mediană a fost de 17,2 luni pentru brațul cu tislelizumab și 11,9 luni pentru brațul cu docetaxel. Perioadele mediane de urmărire pe baza metodologiei Kaplan-Meier inverse au fost de 19,5 luni în brațul cu tislelizumab și de 17,0 luni în brațul cu docetaxel. La finalul analizei (data centralizării datelor 15 iulie 2021), ameliorarea semnificativă clinic a SG a fost observată la setul de analiză a PD-L1 ≥25% care favorizează brațul cu tislelizumab (RR = 0,53; 95% ÎI: 0,41, 0,70; p <0,0001), SG mediană fiind de 19,3 luni pentru brațul cu tislelizumab și 11,5 luni pentru brațul cu docetaxel. Perioada mediană a urmăririi pe baza metodologiei Kaplan-Meier la analiza finală a fost de 31,1 luni în brațul cu tislelizumab și de 27,9 luni în brațul cu docetaxel.
Analiza finală (data centralizării datelor de 15 iulie 2021) a evidențiat rezultate privind eficacitatea în rândul populației ITT constante, comparabile cu cele ale analizei interimare.
Tabelul 6 și Figura 7 sintetizează rezultatele privind eficacitatea în cadrul studiului BGB-A317-303 (set de analiză ITT) la analiza finală.
Tabelul 6 Rezultate privind eficacitatea în cadrul studiului BGB-A317-303
| Obiectiv final | Tislelizumab (N = 535) | Docetaxel (N = 270) |
|---|---|---|
| SG | ||
| Decese, n (%) | 365 (68,2) | 206 (76,3) |
| SG mediană (luni) (IÎ 95%) | 16,9 (15,24, 19,09) | 11,9 (9,63, 13,54) |
| Risc relativ (IÎ 95%)a, b | 0,66 (0,56, 0,79) | |
| SFP | ||
| Evenimente, n (%) | 451 (84,3) | 208 (77,0) |
| SFP mediană (luni) (IÎ 95%) | 4,2 (3,88, 5,52) | 2,6 (2,17, 3,78) |
| Risc relativa (IÎ 95%) | 0,63 (0,53, 0,75) | |
| RRO (%) (IÎ 95%)c | 20,9 (17,56, 24,63) | 3,7 (1,79, 6,71) |
| DRc | ||
| DR mediană (luni) (IÎ 95%) | 14,7 (10,55, 21,78) | 6,2 (4,11, 8,31) |
SG = supraviețuire generală; IÎ = interval de încredere; SFP = supraviețuire fără progresia bolii; RRO = rata răspunsului obiectiv; DR = durata răspunsului. Medianele au fost estimate prin metoda Kaplan-Meier, cu IÎ 95% estimat utilizând metoda Brookmeyer and Crowley. a Riscul relativ a fost estimat din modelul stratificat Cox, cu grupul în care s-a administrat docetaxel ca grup de referință. b Stratificat conform factorilor de stratificare: histologie (scuamos față de non-scuamos), linii de terapie (de a doua intenție față de a treia intenție) și expresia PD-L1 la nivelul celulelor tumorale (scor PD-L1 ≥25% față de scor PD-L1 <25%). c Confirmat de investigator. | ||
Figura 7 Grafic Kaplan-Meier pentru SG în cadrul studiului BGB-A317-303 (setul de analiză ITT)
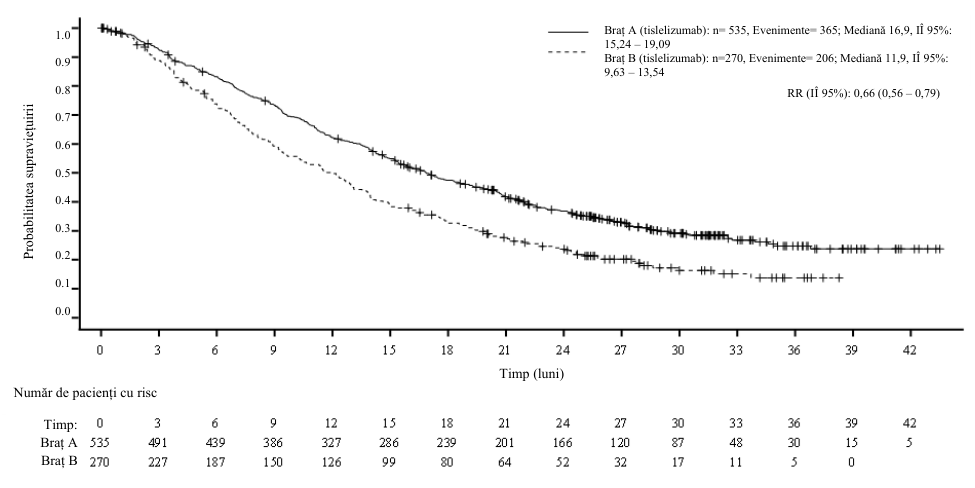
Analizele de subgrup prespecificate au demonstrat un efect constant al tratamentului privind SG în favoarea tislelizumab în subgrupurile majore demografice și de prognostic.
Tabelul 7 sintetizează rezultatele privind eficacitatea pentru SG în funcție de expresia tumorală a PD-L1 (<25% TC, ≥25% TC) în analizele de subgrup prespecificate.
Tabelul 7 Rezultatele privind eficacitatea prin prisma SG în funcție de expresia tumorală a
PD-L1 (<25% TC, ≥25% TC) în cadrul studiului BGB-A317-303
| Brațul cu tislelizumab | Brațul cu docetaxel | |
| N = 535 | N = 270 | |
| Expresia PD-L1 în celule tumorale <25%, n | 307 | 152 |
| Evenimente, n (%) | 223 (72,6) | 117 (77,0) |
| SG mediană (luni) (IÎ 95%) | 15,2 (13,4, 17,6) | 12,3 (9,3, 14,3) |
| Risc relativa (IÎ 95%) | 0,79 (0,64, 0,99) | |
| Expresia PD-L1 în celule tumorale ≥25%, n | 227 | 115 |
| Evenimente, n (%) | 141 (62,1) | 86 (74,8) |
| SG mediană (luni) (IÎ 95%) | 19,3 (16,5, 22,6) | 11,5 (8,2, 13,5) |
| Risc relativa (IÎ 95%) | 0,54 (0,41, 0,71) | |
| a Risc relativ și IÎ 95% asociat au fost estimate din modelul nestratificat Cox. | ||
|---|---|---|
Cancer pulmonar cu celule mici
Tratamentul de primă linie al SCLC în stadiu extins: BGB-A317-312
BGB-A317-312 a fost un studiu randomizat, în regim dublu-orb, multicentric, de fază 3, pentru a compara eficacitatea și siguranța tislelizumab în asociere cu cisplatină sau carboplatină plus etopozidă versus placebo în asociere cu cisplatină sau carboplatină plus etopozidă ca tratament de primă linie la pacienții cu cancer pulmonar cu celule mici în stadiu extins (ES-SCLC).
Studiul a inclus pacienți cu diagnostic confirmat histologic sau citologic de ES-SCLC cărora nu li s-a administrat niciun tratament sistemic anterior pentru ES-SCLC și status de performanță ECOG 0 sau 1.
În total 457 de pacienți au fost randomizați (1:1) pentru a li se administra:
- Brațul cu tislelizumab + chimioterapie: tislelizumab 200 mg plus ASC carboplatină 5 mg/ml/min sau cisplatină 75 mg/m2 în ziua 1 și etopozidă 100 mg/m2 intravenos în zilele 1, 2 și 3 ale fiecărui ciclu de 21 de zile timp de maximum 4 cicluri.
- Brațul cu placebo + chimioterapie: placebo plus carboplatină ASC 5 mg/ml/min sau cisplatină 75 mg/m2 în ziua 1 și etopozidă 100 mg/m2 intravenos în zilele 1, 2 și 3 ale fiecărui ciclu de 21 de zile timp de maximum 4 cicluri.
Alegerea agentului cu platină (cisplatină sau carboplatină) a fost la latitudinea investigatorului. Tislelizumab 200 mg în monoterapie sau placebo a continuat la fiecare 3 săptămâni până la progresia bolii, pierderea beneficiului clinic, toxicitate inacceptabilă.
Randomizarea a fost stratificată în funcție de statutul de performanță ECOG (0 versus 1), chimioterapia aleasă de investigator (carboplatină versus cisplatină) și metastazele cerebrale (da versus nu).
Criteriul final de evaluare principal a fost supraviețuirea globală (SG) în setul de analiză cu intenția de tratament. Criteriile de evaluare finale secundare de eficacitate au inclus supraviețuirea fără progresie (SFP) evaluată de investigator, rata de răspuns obiectiv (RRO) și durata răspunsului (DoR) conform RECIST v1.1.
Datele demografice și caracteristicile inițiale au fost în general echilibrate între cele 2 brațe de tratament. Caracteristicile inițiale pentru toți cei 457 de pacienți randomizați au fost: vârsta mediană de 62 de ani (interval: 31 până la 78 de ani); 37,2% aveau vârsta de ≥65 de ani; 81,4% bărbați; 100% asiatici (toți înrolați în China), 84,9% cu ECOG PS de 1; 1,1% au avut antecedente de metastaze cerebrale; 79% au primit carboplatină la alegerea investigatorului; 62,6% erau fumători actuali; și 89,3% aveau boala în stadiul IV definit de AJCC ediția a 7-a.
La momentul analizei finale prespecificate (data limită 19 aprilie 2023), BGB-A317-312 a demonstrat o îmbunătățire semnificativă statistic a SG pentru pacienții randomizați la brațul cu tislelizumab plus chimioterapie comparativ cu brațul cu placebo plus chimioterapie. RR stratificat a fost de 0,75 (IÎ 95%: 0,61, 0,93; valoarea p unilaterală față de 0,004), cu o SG mediană de 15,5 luni în brațul cu tislelizumab plus chimioterapie, comparativ cu 13,5 luni în brațul cu placebo plus chimioterapie.
O analiză descriptivă actualizată (data limită 29 decembrie 2023) a indicat rezultate consistente în materie de eficacitate cu analiza finală. Timpii mediani de urmărire a SG prin metodologia Kaplan- Meier inversă au fost de 39,8 luni (IÎ 95%: 36,2 până la 41,4 luni) în brațul de tratament cu tislelizumab plus chimioterapie și de 36,4 luni (IÎ 95%: 35,0 până la 40,9 luni) în brațul placebo plus chimioterapie.
Rezultatele privind eficacitatea analizei actualizate sunt prezentate în Tabelul 8 și în Figura 8. Datele pentru pacienții cu metastaze cerebrale sunt prea limitate pentru a trage concluzii despre această populație.
Tabelul 8 Rezultate de eficacitate în BGB-A317-312 – Analiză actualizată
| Tislelizumab + Chimioterapie (N = 227) | Placebo + Chimioterapie (N = 230) | |
| Supraviețuirea globală | ||
|---|---|---|
| Decese, n (%) | 175 (77,1) | 195 (84,8) |
| Mediana (luni) (IÎ 95%)a | 15,5 (13,5, 17,1) | 13,5 (12,1, 14,9) |
| Rata de risc stratificată (IÎ 95%)b | 0,78 (0,63, 0,95) | |
| Supraviețuire fără progresie | ||
| Evenimente, n (%) | 178 (78,4) | 207 (90,0) |
| Mediana (luni) (IÎ 95%)a | 4,7 (4,3, 5,5) | 4,3 (4,2, 4,4) |
| Rata de risc stratificată (IÎ 95%)b | 0,65 (0,53, 0,80) | |
| Rata generală de răspunsc, (%) (IÎ 95%)d | 68,3 (61,8, 74,3) | 61,7 (55,1, 68,0) |
| Durata mediană a răspunsului (luni)c (IÎ 95%)a | 4,3 (4,1, 5,6) | 3,7 (3,0, 4,1) |
a Mediana a fost estimată folosind metoda Kaplan-Meier, cu IÎ 95% estimat folosind metoda Brookmeyer și Crowley cu transformare logaritmică.
b Rata de risc și IÎ 95% au fost estimate folosind un model de regresie Cox stratificat după performanța ECOG (1 vs 0) și platină (carboplatină vs cisplatina) cu placebo + chimioterapie ca grup de referință.
c Răspunsurile obiective au fost confirmate conform RECIST v1.1.
d IÎ 95% a fost estimat folosind metoda Clopper-Pearsonthod.
Figura 8: Graficul Kaplan-Meier a SG în BGB-A317-312
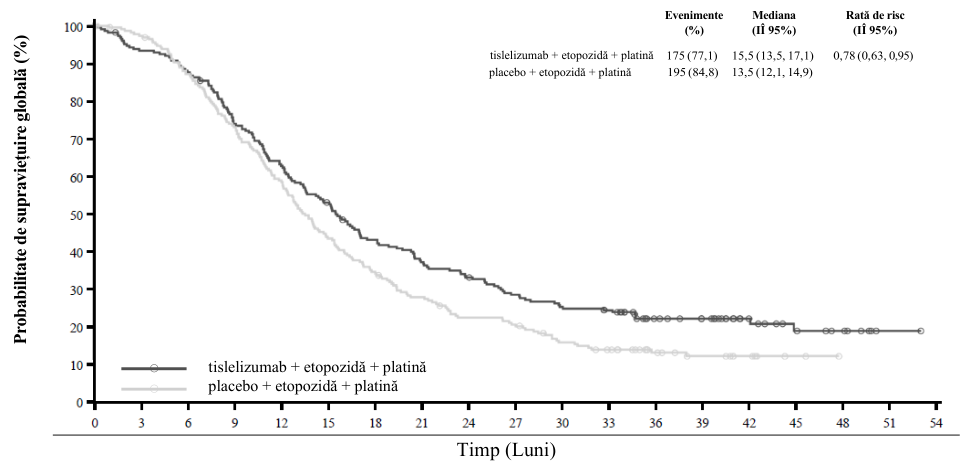
Număr expuși riscului:
tislelizumab
+ etopozidă 227 211 198 166 141 118 95 82 73 62 55 51 33 28 16 10 7 1 0 + platină placebo + etopozidă 230 221 197 165 132 98 78 62 49 45 33 27 17 12 7 2 0 0 0 + platină
Adenocarcinom gastric sau de joncțiune esogastrică (G/JEG)
Tratamentul de primă linie al adenocarcinomului G/GEJ: BGB-A317-305
BGB-A317-305 este un studiu de fază 3 randomizat, multicentric, în regim dublu-orb, controlat cu placebo, care compară eficacitatea și siguranța tislelizumab plus platină și chimioterapie pe bază de fluoropirimidină versus placebo plus chimioterapie pe bază de platină și fluoropirimidină ca tratament de primă linie la pacienții cu adenocarcinom G/JEG local avansat nerezecabil sau metastatic.
Studiul a inclus numai pacienți cu adenocarcinom confirmat histologic și fără terapie sistemică anterioară pentru boala avansată. Este posibil ca pacienții să fi primit anterior terapie neoadjuvantă sau adjuvantă atât timp cât aceasta a fost finalizată și nu au prezenat o recidivă sau progresie a bolii timp de cel puțin 6 luni.
Pacienții au fost înrolați indiferent de nivelul lor de expresie tumorală a PD-L1, care a fost evaluat prospectiv la un laborator central prin scorul de pozitivitate a zonei tumorale (TAP), care este definit ca procentul total din zona tumorală (tumoră și orice stromă desmoplastică) acoperită de celule tumorale cu colorație a membranei PD-L1 (orice intensitate) și celule imune asociate tumorii cu colorație PD-L1 (orice intensitate), estimat vizual de medici anatomo-patologi folosind testul Ventana PD-L1 (SP263).
Studiul a exclus pacienții care aveau cancer cu celule scuamoase sau nediferențiat sau alt tip histologic de tip G/JEG și pacienții care aveau tumori cunoscute HER-2 pozitive.
Randomizarea a fost stratificată în funcție de regiunea geografică (China [inclusiv Taiwan] versus Japonia și Coreea de Sud față de restul lumii [ROW, inclusiv SUA și Europa]), expresia PD-L1 (scorul TAP ≥5% față de scorul TAP <5%), prezența metastazelor peritoneale (da versus nu) și opțiunea ICC (oxaliplatină plus capecitabină versus cisplatină plus 5-FU).
Pacienții au fost randomizați (raport 1:1) pentru a li se administra tislelizumab 200 mg sau placebo la fiecare 3 săptămâni în asociere cu chimioterapie pe bază de platină și fluoropirimidină într-un ciclu de 21 de zile. Tislelizumab (sau placebo) a fost administrat până la progresia bolii sau toxicitate inacceptabilă. După 24 de luni de tratament, terapia studiului poate fi continuată mai mult de doi ani dacă investigatorul consideră că acest lucru este în interesul pacientului pe baza unei evaluări a beneficiului clinic și a riscurilor potențiale.
Chimioterapia a constat din:
- oxaliplatină 130 mg/m² i.v. în ziua 1 și capecitabină 1 000 mg/m2 pe cale orală de două ori pe zi timp de 14 zile consecutive, repetate la fiecare 3 săptămâni. Oxaliplatina a fost administrată timp de până la 6 cicluri, iar capecitabina a fost administrată ca terapie de întreținere la latitudinea investigatorului până la progresia bolii sau toxicitate inacceptabilă. sau
- cisplatină 80 mg/m² i.v. în ziua 1 și 5-FU 800 mg/m2/zi prin perfuzie intravenoasă continuă timp de 24 de ore pe zi în zilele 1-5, repetată la fiecare 3 săptămâni. Cisplatina și 5-FU au fost administrate timp de până la 6 cicluri.
Criteriile finale principale de eficacitate au fost supraviețuirea globală (SG) în setul de analiză pozitivă PD-L1 (scor TAP ≥5%) și setul de analiză ITT (toți pacienții randomizați). Criteriile finale secundare de eficacitate au fost SFP, RRO și DoR, așa cum au fost evaluate de investigator conform RECIST v1.1, și calitatea vieții legate de starea de sănătate (HRQoL).
Evaluarea tumorii a fost efectuată aproximativ la fiecare 6 săptămâni în primele 48 de săptămâni și apoi aproximativ la fiecare 9 săptămâni.
Un total de 997 de pacienți au fost randomizați fie în brațul tislelizumab + chimioterapie (n = 501), fie în brațul placebo + chimioterapie (n = 496). Dintre cei 997 pacienți, 546 (54,8%) au avut scorul TAP ≥5% (tislelizumab + chimioterapie: n = 274; placebo + chimioterapie: n = 272), 931 (93,4%) au primit tratament cu oxaliplatină + capecitabină (tislelizumab + chimioterapie: n = 466; placebo + chimioterapie: n = 465).
La pacienții ale căror tumori au epxrimat PD-L1 cu un scor TAP ≥5%, caracteristicile inițiale pentru populația studiului au fost: vârsta mediană de 62 de ani (interval: 23 până la 84), 39,2% vârsta de 65 de ani sau mai mult; 72,2% bărbați; 23,1% caucazieni și 73,8% asiatici; 33,7% cu ECOG PS de 0 și 66,3% cu ECOG PS de 1. Un total de 79,9% pacienți au avut localizarea tumorală primară a stomacului; 98,5% dintre pacienți au avut boală metastatică la momentul inițial; 43,6% și 39,7% dintre pacienți au avut metastaze hepatice și, respectiv, metastaze peritoneale.
La analiza intermediară prespecificată, BGB-A317-305 a demonstrat o îmbunătățire semnificativă statistic a SG pentru pacienții randomizați la brațul tislelizumab + chimioterapie comparativ cu brațul placebo + chimioterapie la pacienții cu scor TAP ≥5%. IR stratificat a fost de 0,74 (IÎ 95%: 0,59 până la 0,94; valoarea p unilaterală de 0,0056), cu o SG mediană de 17,2 luni în brațul tislelizumab + chimioterapie, comparativ cu 12,6 luni în brațul placebo + chimioterapie. Studiul a demonstrat, de asemenea, o îmbunătățire semnificativă statistic a SFP la pacienții cu scor TAP ≥5%. IR stratificat a fost de 0,67 (IÎ 95%: 0,55 până la 0,83; valoarea p unilaterală < 0,0001), cu o SSP mediană de 7,2 luni pentru tislelizumab + chimioterapie, comparativ cu 5,9 luni pentru placebo + chimioterapie.
La analiza finală prespecificată, BGB-A317-305 a demonstrat o îmbunătățire semnificativă statistic pentru toți pacienții randomizați. IR stratificat a fost de 0,80 (IÎ 95%: 0,70 până la 0,92; valoarea p unilaterală de 0,0011), cu o SG mediană de 15,0 luni în brațul tislelizumab + chimioterapie, comparativ cu 12,9 luni în brațul placebo + chimioterapie. Rezultatele actualizate ale SG la pacienții cu scor TAP ≥5% au fost în concordanță cu rezultatele analizei primare.
Rezultatele finale ale eficacității analizei de la pacienții cu scor TAP ≥5% sunt prezentate în Tabelul 9 și în Figura 9.
Tabelul 9 Rezultate de eficacitate la pacienții cu BGB-A317-305 cu scor TAP ≥5% (analiză finală)
| Tislelizumab + chimioterapie (n = 274) | Placebo + chimioterapie (n = 272) | |
| Pacienți cu scor PD-L1 ≥5% | ||
| Urmărirea mediană a studiului (luni)a | 32,5 | 32,2 |
| SG | ||
|---|---|---|
| Deces, n (%) | 192 (70,1) | 219 (80,5) |
| Medianab (luni) (IÎ 95%) | 16,4 (13,6, 19,1) | 12,8 (12,0, 14,5) |
| Indicele de riscc (IÎ 95%) | 0,71 (0,58, 0,86) | |
| Valoare pc,d | 0,0003e | |
| SFP | ||
| Progresia bolii sau deces, n (%) | 189 (69,0) | 216 (79,4) |
| Medianab (luni) (IÎ 95%) | 7,2 (5,8, 8,4) | 5,9 (5,6, 7,0) |
| Indicele de riscc (IÎ 95%) | 0,68 (0,56, 0,83) | |
| RRO (%) (IÎ 95%) | 51,5 (45,4, 57,5) | 42,6 (36,7, 48,8) |
SG = supraviețuire globală; IÎ = interval de încredere; SFP = supraviețuire fără progresie; RRO = rată de răspuns obiectiv.
a Timpul median de urmărire a fost estimat prin metoda inversă Kaplan-Meier. b Medianele au fost estimate folosind metoda Kaplan-Meier cu IÎ 95% estimate folosind metoda Brookmeyer și Crowley. c Stratificat pe regiuni (Asia de Est versus SUA, Europa) și metastaze peritoneale. d Valoarea p unilaterală din testul log-rank stratificat. e Valoarea nominală p.
Figura 9 Graficul Kaplan-Meier al SG la pacienții cu BGB-A317-305 cu scor TAP ≥5% (analiză finală)
1,0
T+C: n = 274, evenimente = 192, mediana: 16,4, IÎ 95% 13,6, 19,1) Arm A : n = 274 , events = 192 , Median:16.4 , 95% CI 13.6 - 19.1 P+C: n = 272, evenimente = 219, mediana: 12,8, IÎ 95% 12,0, 14,5) Arm B : n = 272 , events = 219 , Median:12.8 , 95% CI 12.0 - 14.5 HR (95% CI) : 0.71 (0.58 - 0.86) IR (IÎ 95%): 0,71 (0,58, 0,86) | ||||||||||||||||||||||
1.0
0,9 0.9
0,8 0.8
0,7 0.7
Survival Probability
0,6 0.6
Probabilitea de supraviețuire
0,5 0.5
0,4 0.4
0,3 0.3
0,2 0.2
0,1 0.1
0.0
0,0
0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 32 34 36 38 40 42 44 46 48 50 Durată (luni) Time (Months)
Număr de pacienți expuși riscului Number of Patients at Risk:
Time: Durată 0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 32 34 36 38 40 42 44 46 48 50 Arm A T+C 274 263 247 228 199 178 156 145 133 120 109 102 97 84 68 50 38 34 27 19 14 9 7 3 1 0 Arm B P+C 272 261 236 215 190 168 148 120 99 83 69 59 53 51 39 29 23 16 14 9 7 3 2 1 0 0
T+C = Tislelizumab + chimioterapie, P+C = placebo + chimioterapie
Atât modelul de rang logaritmic, cât și modelul de regresie Cox au fost stratificate pe regiuni (Asia de Est versus SUA, Europa) și prezența metastazelor peritoneale.
Carcinom esofagian cu celule scuamoase (OSCC)
Tratamentul de primă linie al OSCC: BGB-A317-306
BGB-A317-306 este un studiu, randomizat, în regim dublu-orb, controlat cu placebo, global, de fază III, desfășurat pentru a compara eficacitatea tislelizumabului în asociere cu chimioterapie pe bază de platină versus placebo în asociere cu chimioterapie pe bază de platină la pacienții cu OSCC nerezecabil, local avansat, recurent sau metastatic.
În studiu au fost înrolați pacienți care nu au fost supuși chimioterapiei sau chirurgiei cu intenție curativă. Pacienții au fost înrolați indiferent de nivelul de expresie tumorală a PD-L1. În situațiile în care au fost disponibile, specimenele de țesut tumoral existent/recent prelevat au fost testate retrospectiv pentru a se identifica statusul expresiei PD-L1. Expresia PD-L1 a fost evaluată folosind scorul TAP (pozitivitatea ariei tumorale), definit ca procentul total al zonei tumorale (tumora și orice strom desmoplastic) acoperit de celule tumorale cu colorare a membranei PD-L1 la orice intensitate și de celule imune asociate tumorii cu colorație PD-L1 la orice intensitate, așa cum a fost estimat vizual utilizând testul VENTANA PD-L1 (SP263).
Pacienții care au primit anterior tratament sistemic pentru boala avansată sau metastatică au fost excluși. A fost necesar un interval fără tratament de cel puțin 6 luni dacă pacientul a primit anterior terapie neoadjuvantă/adjuvantă cu chimioterapie pe bază de platină.
Studiul a exclus pacienții care prezentau dovezi de fistulă sau obstrucție esofagiană completă care nu putea fi supusă tratamentului.
Randomizarea a fost stratificată în funcție de regiunea geografică (Asia [cu excepția Japoniei] versus Japonia versus restul lumii [ROW]), terapia definitivă anterioară (da versus nu) și chimioterapia aleasă de investigator (ICC; platină cu fluoropirimidină sau platină cu paclitaxel).
Pacienții au fost randomizați (1:1) pentru a li se administra tislelizumab 200 mg sau placebo la fiecare 3 săptămâni în asociere cu chimioterapia aleasă de investigator (ICC) pe un ciclu de 21 de zile. Regimul dublet de chimioterapie a constat din:
- platină (cisplatină [60 până la 80 mg/m2 i.v. în ziua 1] sau oxaliplatină [130 mg/m2 i.v. în ziua 1]) și o fluoropirimidină (5-FU [750 până la 800 mg/m2 i.v. în zilele 1-5] sau capecitabină [1000 mg/m2 oral de două ori pe zi în zilele 1-4]), sau
- platină (cisplatină [60-80 mg/m2 i.v. în ziua 1 sau 2] sau oxaliplatină [130 mg/m2 i.v. în ziua 1 sau 2]) și paclitaxel (175 mg/m2 i.v. în ziua 1).
Pacienții au fost tratați cu tislelizumab în asociere cu chimioterapie sau placebo în asociere cu chimioterapie până la progresia bolii, conform evaluării de către investigator și conform RECIST versiunea 1.1 sau până la apariția toxicității inacceptabile. După 24 de luni de tratament, terapia de studiu a putut fi continuată peste doi ani dacă investigatorul a considerat că acest lucru este în interesul pacientului, pe baza unei evaluări a beneficiului clinic și a riscurilor potențiale.
Evaluările tumorale au fost efectuate la intervale de 6 săptămâni în primele 48 de săptămâni, apoi la intervale de 9 săptămâni.
Obiectivul final principal privind eficacitatea a fost supraviețuirea generală (SG) la populația cu intenție de tratare (intent to treat - ITT). Obiectivele finale secundare privind eficacitatea au fost supraviețuirea fără progresia bolii (progression-free survival - PFS), rata de răspuns obiectiv (objective response rate - ORR), și durata răspunsului (duration of response - DoR), conform evaluării de către investigator și conform RECIST v1.1, OS în subgrupul PD-L1 pozitiv (scor TAP ≥10%) și calitatea vieții raportată la gradul de sănătate (health-related quality of life - HRQoL).
Un total de 649 pacienți au fost randomizați pentru li se administra tislelizumab în asociere cu chimioterapie (N = 326) sau placebo în asociere cu chimioterapie (N = 323). Dintre cei 649 de pacienți, 290 (44,7%) pacienți au primit platină + fluoropirimidină, 358 de pacienți au avut scor TAP ≥ 5%, 184 de pacienți au avut scor TAP < 5% și 107 pacienți au avut status PD-L1 necunoscut.
La pacienții ale căror tumori au exprimat PD-L1 cu un scor TAP ≥ 5%, caracteristicile inițiale au fost: vârsta mediană 63,0 ani (interval: 40 până la 84), 44,7% vârsta 65 ani sau mai mult; 84,9% bărbați; 20,9% caucazieni și 78,2% asiatici. 87,7% aveau boală metastatică la intrarea în studiu și 12,3% aveau boală local avansată. Toți pacienții au avut confirmarea histologică a carcinomului cu celule scuamoase. Statusul de performanță ECOG inițial a fost 0 (29,9%) sau 1 (70,1%).
De la data-limită a analizei intermediare a datelor (28 februarie 2022), BGB-A317-302 a evidențiat o îmbunătățire semnificativă din punct de vedere statistic a SG în rândul tuturor pacienților. HR stratificat a fost 0,66 (95% CI, 0,54-0,80, valoare p unilaterală < 0,0001), cu o OS mediană de 17,2 luni pentru brațul tislelizumab cu chimioterapie vs. 10,6 luni pentru brațul placebo cu chimioterapie.
O analiză actualizată (urmărire până la 3 ani; data limită a datelor 24 noiembrie 2023) a indicat rezultate de eficacitate consecvente cu analiza intermediară. Timpii mediani de urmărire conform metodologiei inverse Kaplan-Meier au fost de 44,2 luni în brațul tislelizumab în asociere cu chimioterapie și de 43,8 luni în brațul placebo în asociere cu chimioterapie.
Rezultatele privind eficacitatea pentru pacienții cu scor TAP ≥ 5% la o urmărire de 3 ani sunt prezentate în Tabelul 10 și Figura 10.
Tabelul 10 Rezultate privind eficacitatea la pacienții din cadrul studiului BGB-A317-306 cu scor TAP ≥ 5% - Urmărire de 3 ani (data limită a datelor 24 noiembrie 2023)
| Obiectiv final | Tislelizumab + chimioterapie (n = 172) | Placebo + chimioterapie (n = 186) |
|---|---|---|
| SG | ||
| Decese, n (%) | 128 (74,4) | 151 (81,2) |
| Mediană (luni) (IÎ 95%) | 19,1 (16,1; 24,1) | 10,0 (8,6; 11,9) |
| IR (IÎ 95%)a | 0,62 (0,49; 0,79) | |
| valoare pb | < 0,0001 | |
| SFP | ||
| Evenimente, n (%) | 119 (69,2) | 153 (82,3) |
| Mediană (luni) (IÎ 95%) | 8,2 (7,0; 9,8) | 5,5 (4,3; 6,4) |
| valoare pb | < 0,0001 | |
| RRO, (%) (IÎ 95%)c | 64,0 (56,3; 71,1) | 36,0 (29,1; 43,4) |
| SG = supraviețuire generală; IÎ = interval de încredere; IR = indice de risc; SFP = supraviețuire fără progresia bolii; RRO = rata răspunsului obiectiv; a Pe baza unui model de regresie Cox stratificat. b Valoarea p nominală unilaterală pe baza unui test log-rank stratificat. c Intervalul exact de încredere Clopper-Person [sic: Pearson] bilateral. | ||
Figura 10 Grafic Kaplan-Meier privind SG la pacienții din cadrul studiului BGB-A317-306 cu scor TAP ≥ 5% - Urmărire de 3 ani (data limită a datelor 24 noiembrie 2023)
1,0 1.0
| Tislelizumab + Chemotherapy : n = 172 , Events = 128 Median : 19.1 , 95% CI 16.1 - 24.1 Tislelizumab + Chimioterapie: n = 172, Evenimente = 128 Mediană: 19,1, IÎ 95% 16,1 - 24,1 Placebo + Chimioterapie: n = 186, Evenimente = 151 Mediană: 10,0, IÎ 95% 8,6 - 11,9 Placebo + Chemotherapy : n = 186 , Events = 151 Median : 10.0 , 95% CI 8.6 - 11.9 HR (95% CI) : 0.62 (0.49 - 0.79) IR (IÎ 95%): 0,62 (0,49 - 0,79) | ||||||||||||||||||||||||||||||||
0,9 0.9
0,8 0.8
Probabilitatea de supraviețuire
0,7 0.7
Survival Probability
0,6 0.6
0,5
0.5
0,4
0.4
0,3
0.3
0,2
0.2
0,1
0.1
0,0
0.0
0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 32 34 36 38 40 42 44 46 48 50 52 54 56 58 60 Time (Months) Timp (luni)
Număr de pacienți expuși la risc:
Number of Patients at Risk:
Time: Timp: 0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 32 34 36 38 40 42 44 46 48 50 52 54 56 58 60 Tislelizumab + Tislelizumab
172 167 159 146 140 127 116 107 98 86 80 76 70 60 55 50 48 46 40 37 29 25 21 15 12 6 4 3 2 1 0 +Chemotherapy Chimioterapie
Placebo + Placebo
186 181 159 142 113 89 74 69 64 55 50 43 39 35 32 30 28 24 22 22 20 16 13 11 7 5 1 1 0 0 0 +Chemotherapy Chimioterapie
Indicele de risc se bazează pe un model de regresie Cox stratificat.
OSCC tratat anterior: BGB-A317-302
BGB-A317-302 a fost un studiu randomizat, controlat, deschis, global, de fază 3, desfășurat pentru a compara eficacitatea tislelizumab comparativ cu eficacitatea chimioterapiei la pacienții cu OSCC nerezecabil recidivant, în stadiu local avansat sau metastazat, care a progresat pe parcursul administrării prealabile de tratament sistemic sau după aceasta. Pacienții au fost înrolați indiferent de nivelul de expresie tumorală a PD-L1. În situațiile în care au fost disponibile, specimenele de țesut tumoral existent sau recent prelevat au fost testate retrospectiv pentru a se identifica statusul expresiei
PD-L1. Expresia PD-L1 a fost evaluată într-un laborator central, folosind testul Ventana PD-L1 (SP263), care a identificat, printr-o tehnică de colorare, prezența PD-L1, atât la nivelul celulelor tumorale, cât și la nivelul celulelor imune asociate tumorii.
Studiul a exclus pacienții cărora li s-a administrat anterior tratament cu inhibitori anti-PD-1/PD-L1 și care au prezentat invazie tumorală la nivelul organelor adiacente locului de prezentare a bolii la nivel esofagian (de exemplu, aorta sau tractul respirator).
Randomizarea a fost stratificată în funcție de regiunea geografică (Asia [exclusiv Japonia] comparativ cu Japonia comparativ cu SUA/UE), ECOG PS (0 comparativ cu 1) și alegerea opțiunii de chimioterapie de către investigator (investigator choice of chemotherapy - ICC) (paclitaxel comparativ cu docetaxel comparativ cu irinotecan). Alegerea ICC a fost efectuată de investigator înainte de randomizare.
Pacienții au fost randomizați (1:1) pentru a li se administra tislelizumab 200 mg la interval de 3 săptămâni sau chimioterapia aleasă de investigator (ICC), selectată dintre următoarele, toate administrate intravenos:
- paclitaxel 135 până la 175 mg/m² în ziua 1, administrat la interval de 3 săptămâni (și la doze de 80 până la 100 mg/m2 într-un regim săptămânal, conform recomandărilor locale și/sau naționale privind asistența standard), sau
- docetaxel 75 mg/m2 în ziua 1, administrat la interval de 3 săptămâni, sau
- irinotecan 125 mg/m2 în zilele 1 și 8, administrat la interval de 3 săptămâni.
Pacienții au fost tratați cu Tevimbra sau cu una dintre ICC până la progresia bolii, conform evaluării de către investigator și conform RECIST versiunea 1.1 sau până la apariția toxicității inacceptabile.
Evaluările tumorale au fost efectuate la intervale de 6 săptămâni în primele 6 luni, apoi la intervale de 9 săptămâni.
Obiectivul final principal privind eficacitatea a fost supraviețuirea generală (SG) la populația cu intenție de tratare (intent to treat - ITT). Obiectivele finale secundare privind eficacitatea au fost SG în setul de analize cu PD-L1 pozitiv (scor PD-L1 conform Scorului pozitiv combinat estimat vizual, cunoscut acum sub denumirea de scor TAP [Tumour Area Positivity score] PD-L1 ≥10%), rata de răspuns obiectiv (RRO), supraviețuirea fără progresia bolii (SFP) și durata răspunsului (DR), conform evaluării de către investigator și conform RECIST v1.1.
Un total de 512 pacienți au fost înrolați și randomizați pentru li se administra tislelizumab (N = 256) sau ICC (N = 256; paclitaxel [n = 85], docetaxel [n = 53] sau irinotecan [n = 118]). Dintre cei 512 pacienți, 142 (27,7%) au prezentat un scor PD-L1 ≥10%, 222 (43,4%) au prezentat un scor PD-L1 <10% și 148 (28,9%) au prezentat un status inițial PD-L1 necunoscut.
Caracteristicile de bază ale populației în studiu au fost: vârsta mediană de 63 ani (interval: 35 până la 86), 39,5% vârsta de 65 ani sau peste această vârstă; 84% bărbați; 19% caucazieni și 80% asiatici; 25% cu ECOG PS de 0 și 75% cu ECOG PS de 1. Nouăzeci și cinci de procente din populația studiată au prezentat boală în stadiu metastazat la intrarea în studiu. Tuturor pacienților li s-a administrat anterior minimum o schemă de tratament cu chimioterapie antineoplazică, pentru 97% dintre pacienți aceasta reprezentând o schemă dublă de chimioterapie pe bază de platină.
La momentul analizei finale prespecificate, BGB-A317-302 a evidențiat o îmbunătățire semnificativă din punct de vedere statistic a SG în rândul pacienților randomizați în brațul de tratament în care s-a administrat tislelizumab comparativ cu brațul de tratament în care s-a administrat ICC. Riscul relativ stratificat a fost de 0,70 (IÎ 95%: 0,57, 0,85; valoarea p unilaterală 0,0001), cu o valoare mediană a SG de 8,6 luni (IÎ 95%: 7,5, 10,4) în brațul cu tislelizumab în comparație cu 6,3 luni (IÎ 95%: 5,3, 7,0) în brațul cu ICC. Timpii mediani de urmărire conform metodologiei inverse Kaplan-Meier au fost de 20,8 luni în brațul de tratament în care s-a administrat tislelizumab și de 21,1 luni în brațul de tratament în care s-a administrat ICC.
O analiză actualizată cu o urmărire suplimentară de 24 luni după analiza finală prespecificată a indicat rezultate de eficacitate consecvente cu analiza finală. Perioadele mediane de urmărire pe baza metodologiei Kaplan-Meier inverse au fost de 44,7 luni în brațul cu tislelizumab și de 44,0 luni în brațul cu ICC.
Rezultatele privind eficacitatea ale analizei actualizate sunt evidențiate în Tabelul 11 și Figura 11.
Tabelul 11 Rezultate privind eficacitatea în cadrul studiului BGB-A317-302 – Analiza actualizată
| Obiectiv final | Tevimbra (N = 256) | Chimioterapie (N = 256) |
|---|---|---|
| SG | ||
| Decese, n (%) | 233 (91,0) | 233 (91,0) |
| Mediană (luni)a (IÎ 95%) | 8,6 (7,5, 10,4) | 6.3 (5.3, 7,0) |
| Risc relativ (IÎ 95%)b | 0,71 (0,59, 0,86) | |
| valoare pc | P = 0,0002 | |
| SFP evaluată de investigatord | ||
| Progresia bolii sau deces, n (%) | 229 (89,5) | 181 (70,7) |
| Mediană (luni) (IÎ 95%) | 1,6 (1,4, 2,7) | 2,1 (1,5, 2,7) |
| Risc relativ (IÎ 95%) | 0,82 (0,67, 1,01) | |
| RRO cu confirmare de către investigatord | ||
| RRO (%) (IÎ 95%) | 15,2 (11,1, 20,2) | 6,6 (3,9, 10,4) |
| Durata mediană a răspunsului cu confirmarea de către investigator (luni) (IÎ 95%) | 11,3 (6,5, 14,4) | 6,3 (2,8, 8,5) |
| SG = supraviețuire generală; IÎ = interval de încredere; SFP = supraviețuire fără progresia bolii; RRO = rata răspunsului obiectiv; a Estimat conform metodei Kaplan-Meier. b Pe baza modelului de regresie Cox, incluzând tratamentul ca o covariată și stratificat după statusul ECOG inițial și alegerea de către investigator a chimioterapiei. c Valoarea p unilaterală nominală pe baza unui test log-rank unilateral stratificat după statusul de performanță ECOG și alegerea de către investigator a chimioterapiei. d Pe baza analizei ad-hoc. | ||
Figura 11 Grafic Kaplan-Meier privind SG în cadrul studiului BGB-A317-302 (set de analiză ITT) – analiza actualizată
| 1.0 1.0 0.9 | |
|---|---|
| 0.9 Tislelizumab: Tislelizumab : n n = = 256 256, , events evenimente = 197 Median:8.6 = 233 Mediană: ,95% CI 8,6, 7.5 - IÎ 10.4 95% 7,5 – 10,4 0.8 0.8 ICC: n = ICC 256, : evenimente n = 256 , events = 233 = 213 Mediană: Median:6.3 6,3, ,95% IÎ 95% CI 5.3 5,3 - 7.0 – 7,0 | |
| supraviețuirii 0.7 0.7 Probability 0.6 0.6 Probabilitatea 0.5 0.5 Survival 0.4 0.4 0.3 0.3 0.2 0.2 0.1 0.1 0.0 0.0 | HR RR (95% (IÎ 95%): CI) : 0.70(0.57 0,71 (0,59 - 0.85) – 0,86) Valoare Log-rank p a test testului p-value log : rank: 0.0001 0,0001 |
| 0 2 4 6 8 10 12 14 16 | � 18 20 22 24 26 28 30 32 |
1.0
1.0
0.9
0.9
0.8
Probabilitatea supraviețuirii 0.8
0.7
0.7
Survival Probability
0.6
0.6
0.5 0.5
0.4 0.4
0.3 0.3
0.2 0.2
0.1 0.1
0.0
0.0
Timp (luni)
Time (Months)
Număr de pacienți cu risc:
Number of Patients at Risk:
Timp
Time: 0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 Tislelizumab Tislelizumab 256 245 226 214 191 172 157 144 134 122 110 96 88 81 73 63 59 52 44 35 30 25 20 18 13 11 8 8 8 3 2 1 0 ICC ICC 256 235 219 191 167 143 124 105 93 83 77 59 51 42 36 34 29 26 21 19 15 11 7 6 5 4 4 2 2 1 1 0 0 Valoarea p unilaterală nominală se bazează pe un test log-rank stratificat pe baza statusului de performanță ECOG și a alegerii de către investigator a chimioterapiei.
Eficacitate și subgrupuri PD-L1 (analiza actualizată):
La analiza actualizată privind SG în subgrupul pozitiv pentru PD-L1 (scor PD-L1 ≥10%), riscul relativ (RR) stratificat pentru SG a fost 0,54 (IÎ 95%: 0,36 până la 0,79). Supraviețuirea mediană a fost de 10,2 luni (IÎ 95%: 8,5 până la 14,5 luni) și de 5,1 luni (IÎ 95%: 3,8 până la 8,2 luni) pentru brațele de tratament în care s-au administrat tislelizumab, respectiv ICC.
În subgrupul negativ pentru PD-L1 (scor PD-L1 <10%), RR stratificat pentru SG a fost 0,86 (IÎ 95%: 0,65 până la 1,14), cu supraviețuire generală mediană de 7,5 luni (IÎ 95%: 5,5 până la 8,9 luni) și de 5,8 luni (IÎ 95%: 4,8 până la 6,9 luni) pentru brațele de tratament în care s-au administrat tislelizumab, respectiv ICC.
Carcinom nazofaringian (NPC)
Tratamentul de primă linie al NPC recurent sau metastatic: BGB-A317-309
BGB-A317-309 a fost un studiu randomizat, multicentric, în regim dublu-orb, controlat cu placebo, de fază III, pentru compararea eficacității și a siguranței tislelizumabului în asociere cu gemcitabină și cisplatină față de placebo în asociere cu gemcitabină și cisplatină ca tratament de primă linie la pacienții cu NPC recurent sau metastatic.
Pacienții nu au fost tratați anterior pentru NPC recurent sau metastatic. Un interval fără tratament de cel puțin 6 luni a fost necesar dacă pacientului i s-a administrat anterior chimioterapie neoadjuvantă, chimioterapie adjuvantă, radioterapie sau chimioradioterapie cu intenție curativă pentru boală nemetastatică. Studiul a exclus pacienții cu recurență locală adecvată pentru intervenții chirurgicale curative sau cu radioterapie și pacienții cărora li s-au administrat terapii anterioare care țintesc PD-1 sau PD-L1.
Pacienții au fost randomizați (1:1) pentru a li se administra fie tislelizumab 200 mg o dată la 3 săptămâni, fie placebo în asociere cu cisplatină 80 mg/m2 în Ziua 1 plus gemcitabină 1 g/m2 în Ziua 1 și Ziua 8 a fiecărui ciclu de 21 de zile, timp de 4 până la 6 cicluri. Pacienții randomizați au fost stratificați în funcție de sex și de statutul metastazei hepatice.
Tislelizumabul sau placebo a fost administrat până la progresia bolii sau toxicitate inacceptabilă. Pacienților din brațul cu placebo li s-a oferit opțiunea de transfer încrucișat pentru a li se administra tislelizumab în monoterapie după progresia bolii confirmată de IRC.
Criteriul final de evaluare primar privind eficacitatea a fost supraviețuirea fără progresia bolii (SFP), evaluată de către IRC conform criteriilor RECIST v1.1 în setul de analiză al intenției de tratament (ITT). Criteriile finale de evaluare secundare privind eficacitatea au inclus supraviețuirea globală (SG), SFP evaluată de investigator, rata de răspuns obiectiv (RRO) și durata răspunsului (DR), evaluată de IRC.
În total 263 de pacienți au fost randomizați pentru a li se administra fie tislelizumab în asociere cu gemcitabină și cisplatină (N = 131), fie placebo în asociere cu gemcitabină și cisplatină (N = 132).
Caracteristicile inițiale pentru populația de studiu au fost: vârsta mediană de 50 de ani (interval: 23 până la 74 de ani), 91,6% dintre pacienți aveau vârsta sub 65 de ani; 78,3% dintre pacienți erau bărbați; 63,1% aveau un scor SP ECOG de 1; 100% erau asiatici (din China, Thailanda și Taiwan); și 46,7% erau fumători actuali sau foști. 95,1% din populația studiului a avut boală metastatică la randomizare, cu subtipuri histologice de NPC inclusiv 86,3% fără keratinizare, 6,5% carcinom scuamos cheratinizat și 7,2% NPC neclasificat. Majoritatea (76%) pacienților au avut un nivel ADN al virusului Epstein-Barr (EBV) ≥500 UI/ml. Caracteristicile inițiale au fost în general echilibrate între cele 2 brațe.
La momentul analizei intermediare prespecificate (data limită de colectare a datelor 26-mar-2021), BGB-A317-309 a demonstrat o îmbunătățire semnificativă statistic a SFP pentru pacienții randomizați la tislelizumab în asociere cu brațul cu gemcitabină și cisplatină comparativ cu brațul cu placebo plus gemcitabină și cisplatină. IR stratificat a fost de 0,52 (IÎ 95%: 0,38, 0,73; valoarea p unilaterală
<0,0001), cu o SFP mediană de 9,2 luni în brațul cu tislelizumab plus chimioterapie comparativ cu 7,4 luni în brațul cu placebo plus chimioterapie.
O analiză actualizată (data limită de colectare a datelor din 08-dec-2023) a indicat rezultate consecvente privind eficacitatea cu analiza intermediară (Tabelul 12 și Figura 12). În acest moment, 52,3% dintre pacienții din brațul de control au trecut pentru a li se administra tislelizumab în monoterapie. Timpii mediani de urmărire a SG prin metoda Kaplan-Meier inversă au fost de 41,4 luni în brațul cu tislelizumab plus chimioterapie și de 40,8 luni în brațul cu placebo plus chimioterapie.
Datele de la pacienții cu NPC cu vârsta de 65 de ani sau peste sunt prea limitate pentru a trage concluzii la această populație.
Tabelul 12 Rezultatele eficacității în BGB-A317-309 (Setul de analiză ITT) – Analiză actualizată
| Criteriu final deevaluare | Tislelizumab + Chimioterapie (N = 131) | Placebo + Chimioterapie (N = 132) |
|---|---|---|
| SFP stabilită de IRC | ||
| Evenimente, n (%) | 95 (72,5) | 106 (80,3) |
| SFP mediană (luni) (IÎ 95%)a | 9,6 (7,6, 11,6) | 7,4 (5,6, 7,6) |
| Raport de risc stratificat (IÎ 95%)b | 0,53 (0,39, 0,71) | |
| SG | ||
| Decese, n (%) | 55 (42,0) | 64 (48,5) |
| Mediană (luni) (IÎ 95%)a | 45,3 (33,4, NE) | 31,8 (25,0, NE) |
| Raport de risc stratificat (IÎ 95%)b | 0,73 (0,51, 1,05) | |
Abrevieri: NE = neestimabil; SG = supraviețuire globală; IÎ = interval de încredere; SFP = supraviețuire fără progresia bolii.
a Medianele au fost estimate prin metoda Kaplan-Meier cu IÎ 95% estimate folosind metoda Brookmeyer și Crowley. b Stratificat în funcție de sex (bărbat versus femeie) și starea metastazelor hepatice (cu versus fără).
Figura 12 Diagrama Kaplan-Meier a SFP în BGB-A317-309 de către IRC (Setul de analiză
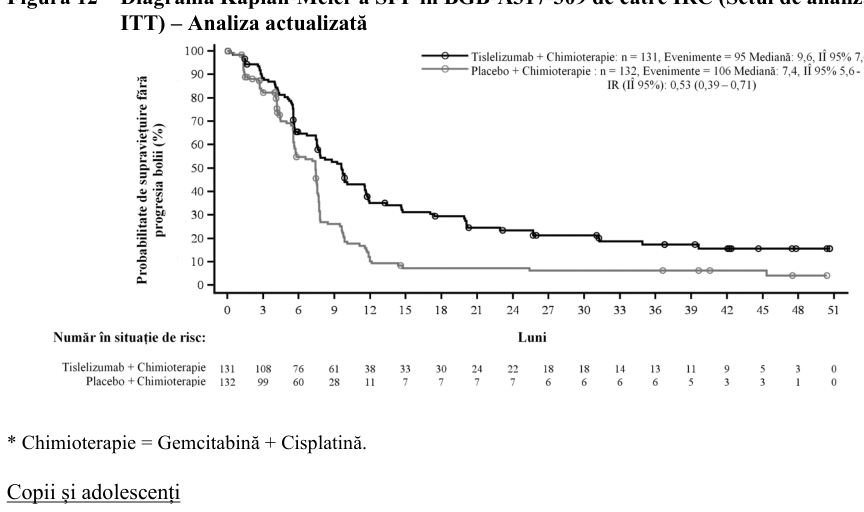
Agenția Europeană pentru Medicamente a acordat o derogare de la obligația de depunere a rezultatelor studiilor efectuate cu tislelizumab la toate subgrupurile de copii și adolescenți în tratamentul neoplaziilor maligne (cu excepția sistemului nervos central, țesut hematopoietic și țesutului limfoid) (vezi pct. 4.2 pentru informații privind utilizarea la copii și adolescenți).
5.2 Proprietăți farmacocinetice
Farmacocinetica (FC) tislelizumab a fost evaluată pentru Tevimbra atât în monoterapie, cât și în asociere cu chimioterapie.
FC tislelizumab a fost caracterizată utilizând analiza farmacocinetică populațională cu date concentrate de la 2596 pacienți, cu neoplazii în stadiu avansat, cărora li s-au administrat doze de tislelizumab de 0,5 până la 10 mg/kg la interval de 2 săptămâni, 2,0 și 5,0 mg/kg greutate corporală la interval de 3 săptămâni și 200 mg la interval de 3 săptămâni.
Perioada de timp până la atingerea nivelului stării de echilibru de 90% este de aproximativ 84 zile (12 săptămâni) după administrarea dozelor de 200 mg la interval de 3 săptămâni și raportul de acumulare la starea de echilibru a expunerii farmacocinetice pentru tislelizumab este aproximativ dublu.
Absorbție
Tislelizumab este administrat intravenos și, prin urmare, este biodisponibil complet și imediat.
Distribuție
O analiză farmacocinetică populațională indică faptul că volumul de distribuție la starea de echilibru este de 6,42 l, valoare specifică anticorpilor monoclonali cu distribuție limitată.
Metabolizare
Se anticipează descompunerea tislelizumab în peptide mici și aminoacizi pe căi catabolice.
Eliminare
Pe baza analizei farmacocinetice populaționale, clearance-ul tislelizumab a fost de 0,153 l/zi, cu o variabilitate interindividuală de 26,3% și media geometrică a timpului de înjumătățire terminal a fost de aproximativ 23,8 zile, cu un coeficient de variație (CV) de 31%.
Liniaritate/Non-liniaritate
La schemele de dozare de 0,5 mg/kg până la 10 mg/kg la interval de 2 sau 3 săptămâni (inclusiv 200 mg la interval de 3 săptămâni și 400 mg o dată la 6 săptămâni), s-a observat că farmacocinetica tislelizumab este liniară și expunerea este proporțională cu doza.
Grupe speciale de pacienți
În analizele farmacocinetice populaționale au fost evaluate efectele diverselor covariate asupra farmacocineticii tislelizumab. Următorii factori nu au avut niciun efect relevant din punct de vedere clinic asupra expunerii la tislelizumab: vârsta (interval 18 până la 90 ani), greutatea corporală (interval 32 până la 130 kg), sexul, rasa (caucazieni, asiatici și alții), insuficiența renală ușoară până la moderată (clearance-ul creatininei [CLCr] ≥30 ml/min), insuficiența hepatică ușoară până la moderată (bilirubină totală ≤3 ori LSN și orice AST) și povara tumorală.
Insuficiență renală
Nu au fost efectuate studii dedicate privind tislelizumab la pacienți cu insuficiență renală. În analizele farmacocinetice populaționale privind tislelizumab, nu au fost identificate diferențe relevante din punct de vedere clinic privind clearance-ul tislelizumab între pacienții cu insuficiență renală ușoară (CLCr 60 până la 89 ml/min, N = 1046) sau insuficiență renală moderată (CLCr 30 până la 59 ml/min, n = 320) și pacienții cu funcție renală normală (CLCr ≥90 ml/min, n = 1223). Insuficiența renală ușoară și moderată nu a avut niciun efect asupra expunerii la tislelizumab (vezi pct. 4.2). Pe baza numărului limitat de pacienți cu insuficiență renală severă (n = 5), efectul insuficienței renale severe asupra farmacocineticii tislelizumab nu este concludent.
Insuficiență hepatică
Nu au fost efectuate studii dedicate privind tislelizumab la pacienți cu insuficiență hepatică. În analizele farmacocinetice populaționale privind tislelizumab, nu au fost identificate diferențe relevante din punct de vedere clinic între pacienții cu insuficiență hepatică ușoară (bilirubină ≤ LSN și AST >LSN sau bilirubină >1,0 până la 1,5 x LSN și orice AST, n = 396) sau insuficiență hepatică moderată (bilirubină >1,5 până la 3 x LSN și orice AST; n = 12), comparativ cu pacienții cu funcție hepatică normală (bilirubină ≤ LSN și AST = LSN, n = 2 182) (vezi pct. 4.2). Pe baza numărului limitat de pacienți cu insuficiență hepatică severă (bilirubină >3 x LSN și orice AST, n = 2), efectul insuficienței hepatice severe asupra farmacocineticii tislelizumab este necunoscut.
5.3 Date preclinice de siguranță
În cadrul studiilor privind toxicitatea la doze repetate la maimuța cynomolgus cu administrare intravenoasă a dozei, la doze de 3, 10, 30 sau 60 mg/kg administrate la interval de 2 săptămâni timp de 13 săptămâni (7 administrări de doze), nu s-au observat nicio toxicitate aparentă legată de tratament sau modificări histopatologice la doze până la 30 mg/kg la interval de 2 săptămâni, corespunzător unei expuneri de 4,3 până la 6,6 ori mai mari decât cele observate la om la doza clinică de 200 mg.
Nu au fost efectuate studii cu tislelizumab la animale privind toxicitatea asupra dezvoltării și reproducerii sau studii privind fertilitatea.
Nu au fost efectuate studii cu tislelizumab pentru a evalua potențialul de carcinogenitate sau genotoxicitate.
6. PROPRIETĂȚI FARMACEUTICE
6.1 Lista excipienților
Citrat de sodiu dihidrat
Acid citric monohidrat
L-histidină clorhidrat monohidrat
L-histidină
Trehaloză dihidrat
Polisorbat 20 (E 432)
Apă pentru preparate injectabile
6.2 Incompatibilități
În absența studiilor de compatibilitate, acest medicament nu trebuie amestecat cu alte medicamente, cu excepția celor menționate la pct 6.6.
6.3 Perioada de valabilitate
Flacon nedeschis
3 ani.
După deschidere
Odată deschis, medicamentul trebuie diluat și perfuzat imediat (vezi pct. 6.6 pentru instrucțiuni privind diluarea medicamentului înainte de administrare).
După prepararea soluției perfuzabile
Tevimbra nu conține conservant. Stabilitatea chimică și fizică în timpul utilizării au fost demonstrate timp de 10 zile (240 ore) la temperaturi de 2 °C până la 8 °C. Cele 10 zile (240 ore) includ păstrarea soluției diluate în condiții de refrigerare (2 °C până la 8 °C), timpul necesar pentru ca soluția să ajungă la temperatura camerei (25 °C sau sub această valoare) și timpul necesar pentru finalizarea perfuzării în decurs de 4 ore.
Din punct de vedere microbiologic, odată diluat, produsul trebuie utilizat imediat.
Dacă nu este utilizat imediat, timpii și condițiile de păstrare în timpul utilizării sunt responsabilitatea utilizatorului. Soluția diluată nu trebuie congelată.
6.4 Precauții speciale pentru păstrare
A se păstra la frigider (2 °C – 8 °C).
A nu se congela.
A se ține flacoanele în cutie pentru a fi protejate de lumină.
Pentru condițiile de păstrare ale medicamentului după diluare, vezi pct. 6.3.
6.5 Natura și conținutul ambalajului
10 ml de concentrat Tevimbra sunt furnizați într-un flacon din sticlă transparentă, de tip 1, cu dop clorobutilic de culoare gri, cu înveliș FluroTec și sigiliu cu buton flip-off.
Tevimbra este disponibil în ambalaje unitare conținând 1 flacon și ambalaje colective conținând 2 (2 ambalaje a câte 1) flacoane.
6.6 Precauții speciale pentru eliminarea reziduurilor și alte instrucțiuni de manipulare
Soluția diluată pentru perfuzare trebuie preparată de către un profesionist din domeniul sănătății, utilizând o tehnică aseptică.
Prepararea soluției pentru perfuzare
- Se scoate numărul de flacoane necesare din frigider, fără a se agita.
- Se inspectează vizual fiecare flacon înainte de administrare pentru a se identifica particule sau decolorare. Concentratul este o soluție limpede până la ușor opalescentă, incoloră până la ușor gălbuie. Nu se utilizează un flacon dacă soluția este tulbure sau dacă se observă particule vizibile sau decolorare.
- Se răstoarnă ușor flacoanele, fără a se agita. Se extrage volumul necesar de soluție din flacon(flacoane) într-o seringă și se transferă într-o pungă de perfuzie intravenoasă care conține soluție injectabilă de clorură de sodiu 9 mg/ml (0,9%) pentru a prepara o soluție diluată cu o concentrație finală cuprinsă între 2 și 5 mg/ml. Se amestecă soluția diluată prin răsturnare ușoară pentru a se evita spumarea sau forfecarea excesivă a soluției.
Administrare
- Se administrează soluția diluată de Tevimbra prin perfuzare printr-o linie de administrare intravenoasă cu un filtru inclus sau adăugat steril, apirogen, cu un nivel scăzut de legare a proteinelor, de 0,2 microni sau 0,22 microni, cu o suprafață de aproximativ 10 cm².
- Pentru doza de 200 mg o dată la 3 săptămâni, prima perfuzie trebuie administrată timp de 60 minute. Dacă este bine tolerată, perfuziile ulterioare pot fi administrate timp de 30 minute.
Perfuzia cu doza inițială de Tevimbra 400 mg trebuie administrată timp de 120 de minute (timp de 90 de minute dacă este utilizată ca tratament ulterior dozei de 200 mg o dată la 3 săptămâni). Dacă este bine tolerată, a doua perfuzie poate fi administrată timp de 60 de minute. Dacă a doua perfuzie este bine tolerată, perfuziile ulterioare pot fi administrate timp de 30 de minute.
- Nu trebuie administrate alte medicamente concomitent prin aceeași linie de perfuzie.
- Tevimbra nu trebuie administrat sub formă de injecție intravenoasă sau injecție unică în bolus.
- Linia intravenoasă trebuie spălată la sfârșitul perfuziei.
- Se elimină orice porțiune neutilizată rămasă în flacon.
- Flacoanele Tevimbra sunt exclusiv de unică folosință.
Eliminare
Orice medicament neutilizat sau material rezidual trebuie eliminat în conformitate cu reglementările locale.
7. DEȚINĂTORUL AUTORIZAȚIEI DE PUNERE PE PIAȚĂ
BeOne Medicines Ireland Limited
10 Earlsfort Terrace
Dublin 2
D02 T380
Irlanda
Tel. +353 1 566 7660
E-mail: beone.ireland@beonemed.com
8. NUMERELE AUTORIZAȚIEI DE PUNERE PE PIAȚĂ
EU/1/23/1758/001-002
9. DATA PRIMEI AUTORIZĂRI SAU A REÎNNOIRII AUTORIZAȚIEI
Data primei autorizări: 15 septembrie 2023
10. DATA REVIZUIRII TEXTULUI
Informații detaliate privind acest medicament sunt disponibile pe site-ul Agenției Europene pentru Medicamente https://www.ema.europa.eu/.