BIXODALAN 500 mg
Rezumatul caracteristicilor produsului (RCP)
- 1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI
- 2. COMPOZIŢIA CALITATIVĂ ŞI CANTITATIVĂ
- 3. FORMA FARMACEUTICĂ
-
4. DATE CLINICE
- 4.1 Indicaţii terapeutice
- 4.2 Doze şi mod de administrare
- 4.3 Contraindicaţii
- 4.4 Atenţionări şi precauţii speciale pentru utilizare
- 4.5 Interacţiuni cu alte medicamente şi alte forme de interacţiune
- 4.6 Fertilitatea, sarcina şi alăptarea
- 4.7 Efecte asupra capacităţii de a conduce vehicule şi de a folosi utilaje
- 4.8 Reacţii adverse
- 4.9 Supradozaj
- 5. PROPRIETĂŢI FARMACOLOGICE
1. DENUMIREA COMERCIALĂ A MEDICAMENTULUI
Bixodalan 500 mg comprimate filmate
2. COMPOZIŢIA CALITATIVĂ ŞI CANTITATIVĂ
Fiecare comprimat filmat conține 500 mg acetat de abirateronă.
Excipienţi cu efect cunoscut
Fiecare comprimat filmat conține 64,6 mg lactoză (68 mg sub formă de monohidrat).
Pentru lista tuturor excipienţilor, vezi pct. 6.1.
3. FORMA FARMACEUTICĂ
Comprimat filmat.
Comprimate filmate, de culoare mov, ovale, marcate cu „500” pe una din feţe, cu dimensiuni de 18,9 mm x 9,5 mm.
4. DATE CLINICE
4.1 Indicaţii terapeutice
Bixodalan este indicat în asociere cu prednison sau prednisolon în:
- tratamentul neoplasmului de prostată, în stadiu metastazic, recent diagnosticat, care răspunde la tratament hormonal (mHSPC - metastatic hormone sensitive prostate cancer), la bărbații adulți, în combinație cu terapie de privare de hormoni androgeni (TPA) (vezi pct. 5.1)
- tratamentul neoplasmului de prostată, în stadiu non-metastazic, cu nivel crescut de risc, recent diagnosticat, care răspunde la tratament hormonal (HSPC- hormone sensitive prostate cancer), la bărbații adulți, în asociere cu TPA și radioterapie (vezi pct. 5.1)
- tratamentul neoplasmului de prostată metastatic, rezistent la orhiectomie (mCRPC - metastatic castration resistant prostate cancer), la bărbaţii adulţi care sunt asimptomatici sau uşor simptomatici, după eşecul terapiei de privare de hormoni androgeni şi la care chimioterapia nu este încă indicată din punct de vedere clinic (vezi pct. 5.1)
- tratamentul neoplasmului de prostată metastatic, rezistent la orhiectomie, la bărbaţii adulţi a căror boală a evoluat în timpul sau după administrarea unei scheme de tratament chimioterapic pe bază pe docetaxel.
4.2 Doze şi mod de administrare
Acest medicament trebuie prescris de un profesionist din domeniul sănătății cu o specializare corespunzătoare.
Doze
Doza recomandată este de 1000 mg (două comprimate de 500 mg) ca doză unică zilnică şi nu trebuie administrată cu alimente (vezi mai jos „Mod de administrare”). Administrarea comprimatelor împreună cu alimentele creşte expunerea sistemică la abirateronă (vezi pct. 4.5 şi 5.2.).
Doza de prednison sau prednisolon
Pentru HSPC în stadiu metastazic și non-metastazic cu nivel crescut de risc, abiraterona este utilizată împreună cu prednison sau prednisolon 5 mg zilnic.
Pentru mCRPC, abiraterona este utilizată împreună cu prednison sau prednisolon 10 mg zilnic.
Durata tratamentului
La pacienţii la care nu s-a efectuat orhiectomie, castrarea medicală cu analogi ai hormonului eliberator de hormon luiteinizant (LHRH) trebuie continuată în timpul tratamentului.
- pentru tratamentul HSPC în stadiu non-metastazic, cu nivel crescut de risc, recent diagnosticat la bărbați adulți, în asociere cu TPA și radioterapie, tratamentul este recomandat până la progresia bolii, toxicitate inacceptabilă sau timp de până la doi ani la pacienții fără progresie a bolii; și
- pentru celelalte indicații tratamentul este recomandat până la progresia bolii sau toxicitate inacceptabilă.
Monitorizare
Concentraţiile serice ale transaminazelor trebuie determinate înainte de iniţierea tratamentului, la interval de două săptămâni, în primele trei luni de tratament şi, ulterior, lunar. De asemenea, trebuie monitorizate lunar tensiunea arterială, potasemia şi retenţia de lichide. Cu toate acestea, pacienţii cu risc major de insuficienţă cardiacă congestivă trebuie monitorizaţi la fiecare 2 săptămâni în timpul primelor trei luni de tratament şi, ulterior, lunar (vezi pct. 4.4).
La pacienţii cu hipopotasemie pre-existentă sau la cei care dezvoltă hipopotasemie în timpul tratamentului cu abirateronă, trebuie avută în vedere menţinerea nivelului de potasiu al pacientului la o valoare ≥ 4,0 mM.
La pacienţii care dezvoltă toxicităţi de Grad ≥ 3 inclusiv hipertensiune arterială, hipopotasemie, edeme şi alte toxicităţi de tip non-mineralocorticoid, tratamentul trebuie întrerupt şi se va institui atitudinea medicală adecvată. Tratamentul cu abirateronă nu trebuie reiniţiat până la remiterea simptomelor toxicităţii la gradul 1 sau la nivelul iniţial. În cazul în care se omite o doză zilnică pentru oricare dintre Bixodalan, prednison sau prednisolon, tratamentul trebuie reluat în ziua următoare, cu doza uzuală zilnică zilnică.
Hepatotoxicitate
La pacienţii care dezvoltă hepatotoxicitate în timpul tratamentului (creşterea concentraţiilor alaninaminotransferazei [ALT] sau creşterea concentraţiilor aspartataminotransferazei [AST] de peste 5 ori faţă de limita superioară a valorilor normale [LSVN]), tratamentul trebuie întrerupt imediat (vezi pct. 4.4). Reluarea tratamentului după revenirea valorilor testelor funcţionale hepatice la valorile iniţiale se poate face cu o doză redusă de 500 mg o dată pe zi. Pentru pacienţii la care se reia tratamentul, concentraţiile serice ale transaminazelor trebuie monitorizate cel puţin la interval de două săptămâni în primele trei luni şi, ulterior, lunar. În cazul în care hepatotoxicitatea reapare la doza redusă de 500 mg pe zi, tratamentul trebuie întrerupt.
Dacă, oricând în timpul tratamentului, pacienţii dezvoltă hepatotoxicitate severă (concentraţii ale ALT sau ale AST de 20 ori mai mari decât LSVN), tratamentul trebuie întrerupt şi nu trebuie reluat.
Insuficienţă hepatică
Nu este necesară ajustarea dozei la pacienţii cu insuficienţă hepatică uşoară preexistentă, clasa A conform clasificării Child-Pugh.
S-a dovedit că prezenţa insuficienţei hepatice moderate (clasa B conform clasificării Child-Pugh) creşte expunerea sistemică la abirateronă de aproximativ 4 ori după administrarea pe cale orală de doze unice de 1000 mg acetat de abirateronă (vezi pct. 5.2). Nu există date privind siguranţa clinică şi eficacitatea administrării de doze multiple de acetat de abirateronă la pacienţii cu insuficienţă hepatică moderată sau severă (clasa B sau C conform clasificării Child-Pugh). Nu se pot face recomandări privind ajustarea dozei. La pacienţii cu insuficienţă hepatică moderată, administrarea abirateronei trebuie evaluată cu atenţie, astfel încât beneficiile să depăşească posibilele riscuri (vezi pct. 4.2 şi 5.2). Abiraterona nu trebuie utilizată la pacienţii cu insuficienţă hepatică severă (vezi pct. 4.3, 4.4 și 5.2).
Insuficienţă renală
Nu este necesară ajustarea dozei pentru pacienţii cu insuficienţă renală (vezi pct. 5.2). Cu toate acestea, nu există experienţă clinică la pacienţii cu neoplasm de prostată şi insuficienţă renală severă. Se recomandă prudenţă la aceşti pacienţi (vezi pct. 4.4).
Copii şi adolescenţi
Utilizarea abirateronei nu este relevantă la copii şi adolescenţi.
Mod de administrare
Bixodalan se administrează pe cale orală.
Comprimatele trebuie administrate cu cel puţin o oră înainte de masă sau la cel puțin două ore după masă. Acestea trebuie înghiţite întregi, cu apă.
4.3 Contraindicaţii
- Hipersensibilitate la substanţa activă sau la oricare dintre excipienţii enumeraţi la pct. 6.1.
- Femei care sunt sau ar putea fi gravide (vezi pct. 4.6).
- Insuficienţă hepatică severă [clasa C conform clasificării Child-Pugh (vezi pct. 4.2, 4.4 şi 5.2)].
- Abiraterona în asociere cu prednison sau prednisolon este contraindicată în combinație cu Ra-223.
4.4 Atenţionări şi precauţii speciale pentru utilizare
Hipertensiune arterială, hipopotasemie, retenţie de lichide şi insuficienţă cardiacă apărută ca urmare a excesului de mineralocorticoizi
Abiraterona poate provoca hipertensiune arterială, hipopotasemie şi retenţie de lichide (vezi pct. 4.8) ca urmare a concentraţiei crescute de mineralocorticoizi ce rezultă din inhibarea CYP17 (vezi pct. 5.1). Administrarea concomitentă a unui corticosteroid inhibă secreţia hormonului adrenocorticotrop (ACTH), determinând reducerea incidenţei şi severităţii acestor reacţii adverse. Este necesară prudenţă în tratamentul pacienţilor a căror afecţiuni medicale preexistente ar putea fi agravate de creşterea tensiunii arteriale, hipopotasemiei (de exemplu pacienţii trataţi cu glicozide cardiotonice) sau de retenţia de lichide (de exemplu, pacienţii cu insuficienţă cardiacă, angină pectorală severă sau instabilă, infarct miocardic recent sau aritmie ventriculară şi cei cu insuficienţă renală severă).
Abiraterona trebuie utilizată cu precauţie la pacienţii cu antecedente de afecţiuni cardiovasculare. Studiile de fază III efectuate cu abirateronă au exclus pacienţii cu hipertensiune arterială necontrolată terapeutic, cu afecţiuni cardiace clinic semnificative, cum sunt infarctul miocardic sau evenimentele trombotice arteriale în ultimele 6 luni, angina pectorală severă sau instabilă, sau insuficienţa cardiacă clasa III sau IV conform New York Heart Association (NYHA) (Studiul 301) sau insuficienţa cardiacă clasa II până la IV (Studiile 3011 și 302) sau cu valori ale fracţiei de ejecţie cardiacă <50%. În Studiile 3011 și 302, au fost excluşi pacienţii cu fibrilaţie atrială sau cu alte aritmii cardiace care necesită tratament medical. Nu a fost stabilită siguranţa la pacienţii cu fracţia de ejecţie a ventriculului stâng (FEVS) <50% sau cu insuficienţă cardiacă clasa III sau IV conform NYHA (în Studiul 301) sau cu insuficienţă cardiacă clasa II până la IV conform NYHA (în Studiile 3011 și 302) (vezi pct. 4.8 şi 5.1).
Înaintea iniţierii tratamentului trebuie luată în considerare obţinerea unei evaluări a funcţiei cardiace (de exemplu, ecocardiogramă) la pacienţii cu risc major de insuficinţă cardiacă congestivă (de exemplu, pacienţi cu istoric de insuficinţă cardiacă, hipertensiune arterială necontrolată, evenimente cardiace cum sunt afecţiunile cardiace ischemice). Înaintea tratamentului cu abirateronă, insuficienţa cardiacă trebuie tratată şi funcţia cardiacă optimizată. Trebuie corectate şi controlate hipertensiunea arterială, hipopotasemia şi retenţia de lichide. În timpul tratamentului, tensiunea arterială, potasemia şi retenţia de lichide (creşterea greutăţii corporale, edeme periferice), dar şi semnele şi simptomele insuficienţei cardiace congestive trebuie monitorizate cel puţin la fiecare 2 săptămâni timp de 3 luni, ulterior, lunar, iar eventualele anomalii trebuie corectate. La pacienții care dezvoltă hipopotasemie în urma tratamentului cu abirateronă, a fost observată prelungirea intervalului QT. În cazul în care se observă o reducere a funcției cardiace, semnificativă din punct de vedere clinic, se evaluează funcția cardiacă așa cum este indicat din punct de vedere clinic, se instituie atitudinea medicală adecvată și se ia în considerare întreruperea acestui tratament (vezi pct. 4.2).
Hepatotoxicitate şi insuficienţă hepatică
În cadrul studiilor clinice controlate au fost observate creşteri semnificative ale valorilor enzimelor hepatice care au determinat întreruperea tratamentului sau modificarea dozei (vezi pct. 4.8). Concentraţiile serice ale transaminazelor hepatice trebuie determinate înainte de iniţierea tratamentului, la interval de două săptămâni în primele trei luni de tratament şi, ulterior, lunar. Dacă apar semne sau simptome clinice care sugerează hepatotoxicitate, trebuie determinate imediat concentraţiile plasmatice ale transaminazelor hepatice. În cazul în care, în orice moment, valorile ALT sau AST cresc de peste 5 ori faţă de LSVN, tratamentul trebuie întrerupt imediat şi funcţia hepatică trebuie monitorizată cu atenţie.
Reluarea tratamentului se poate face numai după revenirea testelor funcţionale hepatice la valorile iniţiale ale pacientului şi numai cu o doză redusă (vezi pct. 4.2).
Dacă oricând în timpul tratamentului pacienţii dezvoltă hepatotoxicitate severă (ALT sau AST de 20 de ori mai mari decât LSVN), tratamentul trebuie întrerupt şi nu trebuie reluat.
Pacienţii cu hepatită virală activă sau simptomatică au fost excluşi din studiile clinice; ca urmare, nu există date care să susţină utilizarea abirateronei la aceşti pacienţi.
Nu există date privind siguranţa clinică şi eficacitatea administrării de doze multiple de acetat de abirateronă la pacienţii cu insuficienţă hepatică moderată sau severă (clasa B sau C conform clasificării Child-Pugh). La pacienţii cu insuficienţă hepatică moderată, administrarea abirateronei trebuie evaluată cu atenţie, astfel încât beneficiile să depăşească posibilele riscuri (vezi pct. 4.2 şi 5.2). Abiraterona nu trebuie utilizată la pacienţii cu insuficienţă hepatică severă (vezi pct. 4.2, 4.3 şi 5.2).
Au fost raportate rar, după punerea pe piaţă, cazuri de insuficienţă hepatică acută şi hepatită fulminantă, unele dintre acestea cu evoluţie letală (vezi pct. 4.8).
Întreruperea administrării corticosteroizilor şi abordarea terapeutică a situaţiilor de stres
Se recomandă prudenţă şi monitorizarea insuficienţei corticosuprarenale care ar putea să apară la pacienţii la care se întrerupe tratamentul cu prednison sau prednisolon. Dacă tratamentul cu abirateronă este continuat după întreruperea administrării corticosteroizilor, pacienţii trebuie monitorizaţi pentru apariţia simptomelor de exces de mineralocorticoizi (vezi informaţiile de mai sus).
La pacienţii trataţi cu prednison sau prednisolon, care sunt expuşi unor situaţii de stres neobişnuite, poate fi indicată creşterea dozei de corticosteroizi înainte, în timpul şi după situaţia stresantă.
Densitate minerală osoasă
Reducerea densităţii minerale osoase poate apărea la bărbaţii cu neoplasm de prostată metastatic în stadiu avansat. Utilizarea abirateronei în asociere cu un glucocorticoid poate accentua acest efect.
Utilizare anterioară de ketoconazol
La pacienţii trataţi anterior cu ketoconazol pentru neoplasm de prostată pot fi anticipate rate de răspuns mai mici.
Hiperglicemie
Utilizarea glucocorticoizilor poate determina hiperglicemie, prin urmare, glicemia trebuie măsurată frecvent la pacienţii cu diabet.
Hipoglicemie
Cazurile de hipoglicemie au fost raportate atunci când abiraterona plus prednison/prednisolon a fost administrată pacienților cu diabet preexistent cărora li s-a administrat pioglitazonă sau repaglinidă (vezi pct. 4.5); prin urmare, glicemia trebuie monitorizată la pacienții cu diabet zaharat.
Utilizare împreună cu chimioterapia
Nu au fost stabilite siguranţa şi eficacitatea utilizării concomitente a abirateronei în asociere cu chimioterapie citotoxică (vezi pct. 5.1).
Intoleranţă la excipienţi
Acest medicament conţine lactoză.
Pacienţii cu afecţiuni ereditare rare de intoleranţă la galactoză, deficit de lactază (Lapp) sau sindrom de malabsorbţie la glucoză-galactoză nu trebuie să utilizeze acest medicament.
Acest medicament conţine sodiu mai puţin de 1 mmol (23 mg) per doză, adică practic „nu conţine sodiu”.
Riscuri potenţiale
Anemia şi disfuncţii sexuale pot apărea la bărbaţii cu neoplasm de prostată metastatic, inclusiv cei aflaţi în tratament cu abirateronă.
Efecte asupra muşchilor scheletici
Au fost raportate cazuri de miopatie și rabdomioliză la pacienţii trataţi cu abirateronă. Majoritatea cazurilor au apărut în perioada primelor 6 luni de tratament şi s-au remis după întreruperea tratamentului cu abirateronă. Se recomandă atenţie la pacienţii trataţi concomitent cu medicamente cunoscute a fi asociate cu miopatie/rabdomioliză.
Interacţiuni cu alte medicamente
În timpul tratamentului se recomandă evitarea inductorilor puternici ai CYP3A4, cu excepţia cazului în care nu există o altă alternativă terapeutică, din cauza riscului de scădere a expunerii la abirateronă (vezi pct. 4.5).
Combinația de abirateronă și prednison/prednisolon cu Ra-223
Tratamentul cu abirateronă și prednison/prednisolon în combinație cu Ra-223 este contraindicat (vezi pct. 4.3) din cauza unui risc crescut de fracturi și a unei tendințe de mortalitate crescută în rândul pacienților cu cancer de prostată asimptomatic sau cu simptome ușoare, așa cum se observă în studiile clinice. Se recomandă ca tratamentul ulterior cu Ra-223 să nu fie inițiat timp de cel puțin 5 zile după ultima administrare a abirateronei în combinație cu prednison/prednisolon.
4.5 Interacţiuni cu alte medicamente şi alte forme de interacţiune
Efectul alimentelor asupra acetatului de abirateronă
Administrarea împreună cu alimente creşte în mod semnificativ absorbţia acetatului de abirateronă. Eficacitatea şi siguranţa în contextul administrării împreună cu alimente nu au fost stabilite, prin urmare, acest medicament nu trebuie administrat împreună cu alimente (vezi pct. 4.2 şi 5.2).
Interacţiuni cu alte medicamente
Potenţialul altor medicamente de a influenţa expunerea la abirateronă
În cadrul unui studiu clinic privind interacţiunile farmacocinetice la subiecţi sănătoşi trataţi anterior cu un inductor puternic al izoenzimei CYP3A4, rifampicina, administrată în doză de 600 mg zilnic timp de 6 zile, urmată de o doză unică de acetat de abirateronă 1000 mg, valoarea plasmatică medie a ASC∞ pentru abirateronă a scăzut cu 55%.
În timpul tratamentului, se recomandă evitarea inductorilor puternici ai izoenzimei CYP3A4 (de exemplu, fenitoină, carbamazepină, rifampicină, rifabutină, rifapentină, fenobarbital, sunătoare
[Hypericum perforatum]), cu excepţia cazului în care nu există o altă alternativă terapeutică.
În cadrul unui studiu clinic separat privind interacţiunile farmacocinetice, efectuat la subiecţi sănătoşi, administrarea concomitentă de ketoconazol, un inhibitor puternic al izoenzimei CYP3A4, nu a avut niciun efect clinic semnificativ asupra farmacocineticii abirateronei.
Potenţialul de influenţare a expunerii la alte medicamente
Abiraterona este un inhibitor al enzimelor hepatice CYP2D6 şi CYP2C8.
Într-un studiu pentru stabilirea efectelor acetatului de abirateronă (în asociere cu prednison) cu o doză unică de dextrometorfan, un substrat pentru CYP2D6, expunerea sistemică (ASC) la dextrometorfan a crescut de aproximativ 2,9 ori. ASC24 h pentru dextrorfan, metabolitul activ al dextrometorfanului, a crescut cu aproximativ 33%.
Se recomandă prudenţă la administrarea în asociere cu medicamente care sunt activate sau metabolizate de către CYP2D6, în special cu medicamente cu indice terapeutic îngust. Trebuie luată în considerare reducerea dozei de medicamente cu indice terapeutic îngust care sunt metabolizate de CYP2D6. Exemple de medicamente metabolizate de izoenzima CYP2D6 includ metoprolol, propranolol, desipramină, venlafaxină, haloperidol, risperidonă, propafenonă, flecainidă, codeină, oxicodonă şi tramadol (ultimele trei medicamente necesită CYP2D6 pentru a forma metaboliţii analgezici activi).
Într-un studiu efectuat la subiecți sănătoși pentru evaluarea interacțiunilor medicamentoase asupra CYP2C8, ASC-ul pioglitazonei a crescut cu 46%, iar ASC-ul pentru M-III și M-IV, metaboliții activi ai pioglitazonei, a scăzut fiecare cu 10% atunci când pioglitazona a fost administrată împreună cu o doză unică de acetat de abirateronă 1000 mg.
Pacienții trebuie monitorizați pentru semne de toxicitate legate de un substrat CYP2C8 cu un indice terapeutic îngust dacă acesta este utilizat concomitent. Exemple de medicamente metabolizate de CYP2C8 includ pioglitazonă și repaglinidă (vezi pct. 4.4).
In vitro, s-a demonstrat că metaboliţii principali abirateronă sulfat şi N-oxidul abirateronei sulfat inhibă transportorul recaptării hepatice OATP1B1 şi astfel poate rezulta o creştere a concentraţiei medicamentelor eliminate de către OATP1B1. Nu sunt disponibile date clinice care să confirme interacţiunea pe baza transportorului.
Utilizare împreună cu medicamente cunoscute a prelungi intervalul QT
Deoarece tratamentul de privare de hormoni androgeni poate prelungi intervalul QT, se recomandă prudență în cazul administrării abirateronei cu medicamente cunoscute a prelungi intervalul QT sau cu medicamente ce pot induce torsada vârfurilor, cum sunt medicamentele antiaritmice din clasa IA (de exemplu, chinidină, disopiramidă) sau din clasa III (de exemplu, amiodaronă, sotalol, dofetilid, ibutilidă) sau metadonă, moxifloxacină, antipsihotice etc.
Utilizarea împreună cu spironolactonă
Spironolactona se leagă de receptorul androgenic şi poate creşte valorile antigenului specific al prostatei (PSA). Nu este recomandată utilizarea împreună cu abirateronă (vezi pct. 5.1).
4.6 Fertilitatea, sarcina şi alăptarea
Femeile aflate la vârsta fertilă
Nu există date la om privind utilizarea abirateronei în timpul sarcinii şi acest medicament nu este destinat utilizării la femeile aflate la vârsta fertilă.
Contracepţia la bărbaţi şi femei
Nu se cunoaşte dacă abiraterona sau metaboliţii săi sunt prezenţi în materialul seminal. Se recomandă folosirea prezervativului dacă pacientul are contact sexual cu o gravidă. Dacă pacientul are contact sexual cu o femeie aflată la vârsta fertilă, este necesară folosirea prezervativului împreună cu o altă metodă contraceptivă eficace. Studiile la animale au arătat toxicitate reproductivă (vezi pct. 5.3).
Sarcina
Abiraterona nu este destinată utilizării la femei şi este contraindicată la gravide sau femeile care pot fi gravide (vezi pct. 4.3 şi 5.3).
Alăptarea
Abiraterona nu este destinată utilizării la femei.
Fertilitatea
Abiraterona a afectat fertilitatea la masculii şi femelele de şobolani, dar aceste efecte au fost complet reversibile (vezi pct. 5.3).
4.7 Efecte asupra capacităţii de a conduce vehicule şi de a folosi utilaje
Abiraterona nu are nicio influenţă sau are influenţă neglijabilă asupra capacităţii de a conduce vehicule și de a folosi utilaje.
4.8 Reacţii adverse
Rezumatul profilului de siguranţă
Într-o analiză a reacțiilor adverse din studiile de faza 3 cu abirateronă, reacțiile adverse observate la ≥10% dintre pacienți au fost edeme periferice, hipopotasemie, hipertensiune arterială, infecţii ale tractului urinar și valori crescute ale alaninaminotransferazei și/sau aspartataminotransferazei.
Alte reacţii adverse importante includ afecţiuni cardiace, hepatotoxicitate, fracturi şi alveolită alergică.
Ca o consecinţă farmacodinamică a mecanismului său de acţiune, abiraterona poate provoca hipertensiune arterială, hipopotasemie şi retenţie de lichide. În studii de fază 3, reacţiile adverse de tip mineralocorticoid anticipate au fost observate mai frecvent la pacienţii trataţi cu acetat de abirateronă comparativ cu pacienţii la care s-a administrat placebo: hipopotasemie 18% față de 8%, hipertensiune arterială 22% față de 16% şi, respectiv retenţie de lichide (edeme periferice) 23% față de 17%. La pacienţii trataţi cu acetat de abirateronă față de pacienții tratați cu placebo: s-au observat hipopotasemie gradele 3 și 4 CTCAE (versiunea 4.0) la 6% față 1% dintre pacienți, hipertensiune arterială gradele 3 şi 4 conform CTCAE (versiunea 4.0) la 7% față de 5% și respectiv retenție de lichide (edem periferic) la 1% față de 1% dintre pacienți.
În general, reacţiile de tip mineralocorticoid au fost tratate medical cu succes. Utilizarea concomitentă a unui corticosteroid reduce incidenţa şi severitatea acestor reacţii adverse (vezi pct. 4.4).
Lista reacţiilor adverse sub formă de tabel
În studiile efectuate la pacienţi cu neoplasm de prostată metastazat, în stadiu avansat, care utilizau un analog LHRH sau care au fost trataţi anterior prin orhiectomie, abiraterona a fost administrată în doză de 1000 mg pe zi în asociere cu doze mici de prednison sau prednisolon (5 sau 10 mg zilnic, în funcție de indicație).
Reacţiile adverse observate în timpul studiilor clinice şi în experienţa de după punerea pe piaţă sunt enumerate mai jos pe categorii de frecvenţă. Categoriile de frecvenţă sunt definite după cum urmează: foarte frecvente (≥ 1/10); frecvente (≥ 1/100 şi < 1/10); mai puţin frecvente (≥ 1/1000 şi < 1/100); rare (≥ 1/10000 şi < 1/1000); foarte rare (< 1/10000) şi cu frecvenţă necunoscută (care nu poate fi estimată din datele disponibile).
În cadrul fiecărei grupe de frecvenţă, reacţiile adverse sunt prezentate în ordinea descrescătoare a gravităţii.
Tabelul 1: Reacţii adverse identificate în studiile clinice şi după punerea pe piaţă
| Aparate, sisteme, organe | Reacții adverse și frecvenţa |
|---|---|
| Infecţii şi infestări | foarte frecvente: infecţii ale tractului urinar |
| frecvente: sepsis | |
| Tulburări ale sistemului imunitar | cu frecvență necunoscută: reacții anafilactice |
| Tulburări endocrine | mai puțin frecvente: insuficienţă suprarenală |
| Tulburări metabolice şi de nutriţie | foarte frecvente: hipopotasemie |
| frecvente: hipertrigliceridemie | |
| Tulburări cardiace | frecvente: insuficienţă cardiacă*, angină pectorală, fibrilaţie atrială, tahicardie |
| mai puțin frecvente: alte aritmii | |
| cu frecvență necunoscută: infarct miocardic, prelungirea intervalului QT (vezi pct. 4.4 și 4.5) | |
| Tulburări vasculare | foarte frecvente: hipertensiune arterială |
| Tulburări respiratorii, toracice şi mediastinale | rare: alveolită alergicăa |
| Tulburări gastro-intestinale | foarte frecvente: diaree |
| frecvente: dispepsie | |
| Tulburări hepatobiliare | foarte frecvente: creşterea concentraţiilor de alaninaminotransferazăși/sau aspartataminotransferazăb |
| rare: hepatită fulminantă, insuficienţă hepatică acută | |
| Afecţiuni cutanate şi ale ţesutului subcutanat | frecvente: erupţii cutanate tranzitorii |
| Tulburări musculo-scheletice şi ale țesutului conjunctiv | mai puțin frecvente: miopatie, rabdomioliză |
| Tulburări renale şi ale căilor urinare | frecvente: hematurie |
| Tulburări generale şi la nivelul locului de administrare | foarte frecvente: edeme periferice |
| Leziuni, intoxicaţii şi complicaţii legate de manevre procedurale | frecvente: fracturi** |
* Insuficienţa cardiacă include şi insuficienţa cardiacă congestivă, disfuncţia ventriculară stângă şi scăderea fracţiei de ejecţie a ventriculului stâng
** Fracturile includ fracturile cauzate de osteoporoză și toate tipurile de fracturi, cu excepţia fracturilor de ordin patologic a Raportări spontane din experienţa de după punerea pe piaţă b Valorile crescute ale alaninaminotransferazei și/sau ale aspartataminotransferazei includ ALT crescut, AST crescut, și funcție hepatică anormală.
Următoarele reacţii adverse de gradul 3 conform CTCAE (versiunea 4.0) au apărut la pacienţii trataţi cu acetat de abirateronă: hipopotasemie 5%; infecţii ale tractului urinar 2%; concentraţii crescute ale alaninaminotransferazei și/sau aspartataminotransferazei 4%; hipertensiune arterială 6%; fracturi 2%; edeme periferice, insuficienţă cardiacă şi fibrilaţie atrială, fiecare câte 1%. Angina pectorală şi hipertrigliceridemia de gradul 3 conform CTCAE (versiunea 4.0) au apărut la <1% dintre pacienți. Infecţii ale tractului urinar, valori crescute ale alaninaminotransferazei și/sau aspartataminotransferazei, hipopotasemie, insuficienţă cardiacă, fibrilații atriale și fracturi de gradul 4 conform CTCAE (versiunea 4.0) au apărut la <1% dintre pacienți.
O incidență mai mare a hipertensiunii arteriale și a hipopotasemiei a fost observată la populația sensibilă la tratamentul cu hormoni (studiul 3011). Hipertensiunea arterială a fost raportată la 36,7% dintre pacienții din populația sensibilă la tratamentul cu hormoni (studiul 3011), comparativ cu 11,8% și 20,2% în studiile 301, respectiv 302.
Hipopotasemia a fost observată la 20,4% dintre pacienții din populația sensibilă la tratamentul cu hormoni (studiul 3011) în comparație cu 19,2% și 14,9% în studiul 301, respectiv 302).
Incidența și severitatea evenimentelor adverse au fost mai mari în subgrupul pacienților cu grad 2 al statusului inițial de performanță ECOG și, de asemenea, la pacienții vârstnici (≥75 de ani).
Descrierea anumitor reacţii adverse
Reacţii cardiovasculare
Cele trei studii de fază 3 au exclus pacienţii cu hipertensiune arterială necontrolată terapeutic, afecţiuni cardiace clinic semnificative, precum infarct miocardic, sau evenimente trombotice arteriale în ultimele 6 luni, angină pectorală severă sau instabilă, sau insuficienţă cardiacă clasa III sau IV conform NYHA (studiul 301) sau insuficienţă cardiacă clasa II până la IV (studiile 3011 și 302) sau cu fracţia de ejecţie < 50%. Toţi pacienţii incluşi în studiu (atât pacienţii trataţi cu substanţa activă, cât şi cei la care s-a administrat placebo) au fost trataţi concomitent cu terapie de privare androgenică, în principal, prin utilizarea de analogi ai LHRH, care a fost asociată cu diabet zaharat, infarct miocardic, accident vascular cerebral şi moarte subită de cauză cardiacă. În studiile de fază III, incidenţa reacţiilor adverse cardiovasculare, la pacienţii la care sa administrat acetat de abirateronă în comparaţie cu pacienţii la care s-a administrat placebo au fost după cum urmează: fibrilații atriale 2,6% față de 2,0%, tahicardie 1,9% față de 1,0%, angină pectorală 1,7% față de 0,8%, insuficiență cardiacă 0,7% față de 0,2% și aritmie 0,7% față de 0,5%.
Hepatotoxicitate
La pacienţii trataţi cu acetat de abirateronă a fost raportată hepatotoxicitate manifestată prin concentraţii crescute ale ALT, AST şi bilirubinei totale. Pe parcursul studiilor clinice de fază 3, la aproximativ 6% dintre pacienţii la care s-a administrat acetat de abirateronă a fost raportată hepatotoxicitate de gradele 3 și 4 (de exemplu, creşterea valorilor testelor ALT sau AST de >5 x LSVN sau creşteri ale bilirubinemiei de >1,5 x LSVN), în mod caracteristic în primele 3 luni după iniţierea tratamentului. În cadrul Studiului clinic 301, hepatotoxicitatea de gradul 3 sau 4 a fost observată la 8,4% dintre pacienții tratați cu abirateronă. Zece pacienți cărora li s-a administrat abirateronă au întrerupt definitiv administrarea din cauza hepatotoxicității; doi pacienți au prezentat hepatotoxicitate de gradul 2, șase pacienți au prezentat hepatotoxicitate de gradul 3, iar doi pacienți au prezentat hepatotoxicitate de gradul 4. Niciun pacient nu a murit din cauza hepatotoxicității în Studiul 3011. În studiile clinice din faza 3, a existat o probabilitate mai mare ca pacienții ale căror valori inițiale ALT sau AST au fost crescute să prezinte creșteri ale valorilor testelor funcției hepatice decât în cazul celor care au avut inițial valorile normale. Când au fost observate creșteri ale ALT sau AST > 5 x LSVN sau creșteri ale bilirubinei > 3 x LSVN, administrarea acetatului de abirateronă a fost întreruptă temporar sau definitiv. În două cazuri au avut loc creșteri semnificative ale valorilor testelor funcției hepatice (vezi pct. 4.4). Acești doi pacienți cu funcție hepatică inițială normală au prezentat creșteri ale valorilor ALT sau AST de la 15 la 40 x LSVN și creșteri ale valorilor bilirubinei de la 2 la 6 x LSVN. La întreruperea tratamentului, ambii pacienți au prezentat normalizarea valorilor testelor funcției hepatice, iar un pacient a fost tratat din nou, fără recidiva creșterii acestor valori. În Studiul 302, au fost observate creșteri ale valorilor ALT sau AST de gradul 3 sau 4 la 35 (6,5%) pacienți tratați cu acetat de abirateronă.
Creşterile concentraţiilor aminotransferazelor s-au remis la toţi pacienţii, cu excepţia a 3 cazuri (2 cu metastaze hepatice multiple de novo şi 1 cu creşterea concentraţiei AST la aproximativ 3 săptămâni după administrarea ultimei doze de acetat de abirateronă). În studiile clinice de fază 3, întreruperile definitive ale tratamentului cauzate de creșterea valorilor ALT și AST sau funcției hepatice anormale au fost raportate la 1,1% dintre pacienții tratați cu acetat de abirateronă și 0,6% dintre pacienții tratați cu placebo; nu s-au raportat decese cauzate de evenimentele de hepatotoxicitate.
În studiile clinice, riscul de hepatotoxicitate a fost atenuat prin excluderea pacienţilor cu hepatită sau cu anomalii semnificative ale valorilor testelor funcţiei hepatice la momentul iniţial. În cadrul Studiului 3011, au fost excluși pacienții cu valori inițiale ALT și AST > 2,5 X LSVN, valori ale bilirubinei > 1,5 X LSVN sau cei cu hepatită virală activă sau simptomatică sau cu boală hepatică cronică; ascită sau tulburări hemoragice secundare disfuncției hepatice. Pacienţii cu valori iniţiale ale ALT şi AST ≥ 2,5 x LSVN în absenţa metastazelor hepatice şi > 5 x LSVN în prezenţa metastazelor hepatice au fost excluşi din studiul 301. Pacienţii cu metastaze hepatice care nu au fost eligibili şi pacienţii cu valori iniţiale ale ALT şi AST ≥ 2,5 x LSVN au fost excluşi din studiul 302. Apariţia valorilor modificate ale testelor funcţionale hepatice la pacienţii care au participat în studiile clinice a fost gestionată prompt prin solicitarea întreruperii tratamentului și permiterea reluării acestuia numai după revenirea la valorile iniţiale ale testelor funcţionale hepatice (vezi pct. 4.2). Tratamentul nu a fost reluat la pacienţii cu creşteri ale ALT sau AST > 20 x LSVN. Siguranţa reluării tratamentului la aceşti pacienţi nu este cunoscută. Mecanismul de apariţie al hepatotoxicităţii nu este înţeles.
Raportarea reacţiilor adverse suspectate
Raportarea reacţiilor adverse suspectate după autorizarea medicamentului este importantă. Acest lucru permite monitorizarea continuă a raportului beneficiu/risc al medicamentului. Profesioniştii din domeniul sănătăţii sunt rugaţi să raporteze orice reacţie adversă suspectată la
Agenţia Naţională a Medicamentului şi a Dispozitivelor Medicale din România
Str. Aviator Sănătescu nr. 48, sector 1
Bucureşti 011478- RO e-mail: adr@anm.ro
Website: www.anm.ro
4.9 Supradozaj
Experienţa cu privire la cazurile de supradozaj cu abirateronă la om este limitată.
Nu există un antidot specific. În caz de supradozaj administrarea trebuie întreruptă şi se vor institui măsuri suportive generale, inclusiv monitorizare pentru apariţia aritmiilor, hipopotasemiei şi a semnelor şi simptomelor retenţiei de lichide. De asemenea, trebuie evaluată funcţia hepatică.
5. PROPRIETĂŢI FARMACOLOGICE
5.1 Proprietăţi farmacodinamice
Grupa farmacoterapeutică: terapie endocrină, alţi antagonişti hormonali şi substanţe înrudite, codul ATC: L02BX03
Mecanism de acţiune
Acetatul de abirateronă este convertit in vivo în abirateronă, un inhibitor al biosintezei hormonilor androgeni. Mai exact, abiraterona inhibă selectiv enzima 17α-hidroxilază/C17,20-liaza (CYP17). Această enzimă este exprimată şi este necesară pentru biosinteza hormonilor androgeni în ţesuturile tumorale din testicule, suprarenale şi prostată. CYP17 catalizează conversia pregnenolonei şi progesteronului la precursorii testosteronului, DHEA şi androstenediona, respectiv prin
17α-hidroxilarea şi clivajul legăturii C17,20. De asemenea, inhibarea CYP17 determină creşterea producţiei de mineralocorticoizi de către glandele suprarenale (vezi pct. 4.4).
Carcinoamele de prostată sensibile la hormoni androgeni răspund la tratamentul care scade concentraţiile de hormoni androgeni. Terapiile de privare androgenică, precum tratamentul cu analogi ai LHRH sau orhiectomia, scad producţia testiculară de hormoni androgeni, dar nu influenţează producţia de hormoni androgeni de către glandele suprarenale sau la nivelul tumorii. Tratamentul cu abirateronă scade concentraţiile serice de testosteron la valori nedetectabile (folosind teste comerciale), atunci când se administrează în asociere cu analogi ai LHRH (sau orhiectomie).
Efecte farmacodinamice
Abiraterona scade concentraţia serică a testosteronului şi a altor hormoni androgeni sub valorile obţinute prin utilizarea de analogi ai LHRH în monoterapie sau prin orhiectomie. Acest efect rezultă din inhibarea selectivă a enzimei CYP17 necesară pentru biosinteza de hormoni androgeni. PSA are rol de biomarker la pacienţii cu neoplasm de prostată. În cadrul unui studiu clinic de fază III la pacienţii la care chimioterapia anterioară cu taxani a eşuat, 38% dintre pacienţii trataţi cu acetat de abirateronă au înregistrat o scădere de minimum 50% din valoarea iniţială a valorilor PSA comparativ cu 10% dintre pacienţii la care s-a administrat placebo.
Eficacitatea şi siguranţa clinică
Eficacitatea a fost stabilită în patru studii clinice randomizate, controlate placebo, multicentrice, de fază 3 (Studiile 3011, 302, 301 și STAMPEDE) la pacienţi cu mHSPC, mCRPC și HSPC în stadiu nonmetastazic, cu nivel crescut de risc. Studiul 3011 (LATITUDE) a inclus pacienți recent diagnosticați (în decurs de 3 luni de la randomizare) cu mHSPC care au prezentat factori de prognostic cu risc ridicat. Prognosticul cu risc ridicat a fost definit ca având cel puțin 2 din următorii 3 factori de risc: (1) scor Gleason ≥ 8; (2) prezența a 3 sau mai multe leziuni la scanarea osoasa; (3) prezența metastazelor viscerale masurabile (cu exceptia bolii ganglionilor limfatici). În brațul activ, abiraterona a fost administrată la o doză de 1000 mg zilnic în combinație cu prednison în doză mică de 5 mg o dată pe zi, în asociere cu TDA (agonist LHRH sau orhiectomie), ceea ce a reprezentat protocolul standard de tratament. Pacienții din brațul de control au primit TDA și placebo atât pentru abirateronă, cât și pentru prednison. Studiul 302 a inclus pacienți cărora nu li s-a administrat docetaxel anterior; în timp ce studiul 301 a inclus pacienți cărora li s-a administrat anterior docetaxel. Pacienții își administrau un analog LHRH sau au suferit anterior orhiectomie. În brațul de tratament activ, abiraterona a fost administrată la o doză de 1000 mg zilnic în combinație cu o doză mică de prednison sau prednisolon 5 mg de două ori pe zi. Pacienții cărora li s-a administrat tratament de control li s-a administrat placebo și prednison sau prednisolon în doză mică 5 mg de două ori pe zi.
STAMPEDE a fost un studiu randomizat, controlat, multicentric, pentru pacienții cu neoplasm de prostată local avansat sau în stadiu metastazic care încep TPA pe termen lung. Studiul a avut un design de platformă multigrup, în mai multe etape, încorporând o componentă de fază 2-3 fără întreruperi. Studiul a evaluat efectele adăugării diferiților agenți, atât ca agenți unici, cât și în asocieri, la tratamentul standard. STAMPEDE a înrolat pacienți cu neoplasm de prostată recent diagnosticat și în stadiu metastazic, cu ganglioni pozitivi sau local avansat cu nivel crescut de risc.
Modificările concentrației plasmatice PSA în mod independent nu garantează întotdeauna beneficii clinice. Prin urmare, în toate studiile s-a recomandat ca pacienții să continue tratamentele de studiu până la îndeplinirea criteriilor de întrerupere definitivă, astfel cum se specifică mai jos pentru fiecare studiu.
În toate studiile, utilizarea spironolactonei nu a fost permisă deoarece spironolactona se leagă de receptorul androgen și poate crește nivelurile PSA.
Neoplasm de prostată, în stadiu metastazic, care răspunde la tratament hormonal (mHSPC)
Studiul 3011 (pacienții cu mHSPC cu risc crescut, recent diagnosticat; studiul LATITUDE)
În studiul 3011, (n=1199) vârsta medie a pacienților înrolați a fost de 67 ani. Numărul de pacienți tratați cu abirateronă în funcție de grupul rasial a fost caucazieni 832 (69,4%), asiatici 246 (20,5%), de rasă neagră sau afro-americani 25 (2,1%), alții 80 (6,7%), necunoscuți/neraportați 13 (1,1%) și indieni americani sau persoane din Alaska 3 (0,3%). Statusul performanței ECOG a fost 0 sau 1 pentru 97% dintre pacienți. Au fost excluși pacienții cu metastaze cerebrale cunoscute, hipertensiune arterială necontrolată, boli cardiace semnificative sau insuficiență cardiacă NYHA II - IV. Au fost excluși pacienții care au fost tratați cu farmacoterapie anterioară, radioterapie sau chirurgie pentru cancer metastatic de prostată, cu excepția celor care au administrat TDA până la 3 luni sau 1 schemă de radioterapie paliativă sau terapie chirurgicală pentru a trata simptomele rezultate din boala metastatică. Obiectivele eficacității co-primare au fost supraviețuirea globală (SG) și supraviețuirea fără progresie radiologică (SFPr). Scorul inițial median al durerii, măsurat prin Scorul de evaluare a intensităţii durerii (BPI-SF), a fost de 2,0 atât în grupurile de tratament, cât și în grupurile placebo. În plus față de măsurile co-primare finale, beneficiul a fost, de asemenea, evaluat folosind timpul pentru eveniment legate de schelet (ELS), timpul până la terapia următoare pentru cancerul de prostată, timpul până la inițierea chimioterapiei, timpul până la progresia durerii și timpul până la progresia PSA. Tratamentul a continuat până la progresia bolii, retragerea consimțământului, apariția toxicității inacceptabile sau deces.
Supraviețuirea fără progresie radiologică a fost definită ca timpul de la randomizare până la apariția progresiei radiografice sau a decesului din orice cauză. Progresia radiologică a inclus progresia identificată prin scanare osoasă (conform PCWG2 modificate) sau progresia leziunilor țesuturilor moi identificată prin CT sau RMN (conform RECIST 1.1).
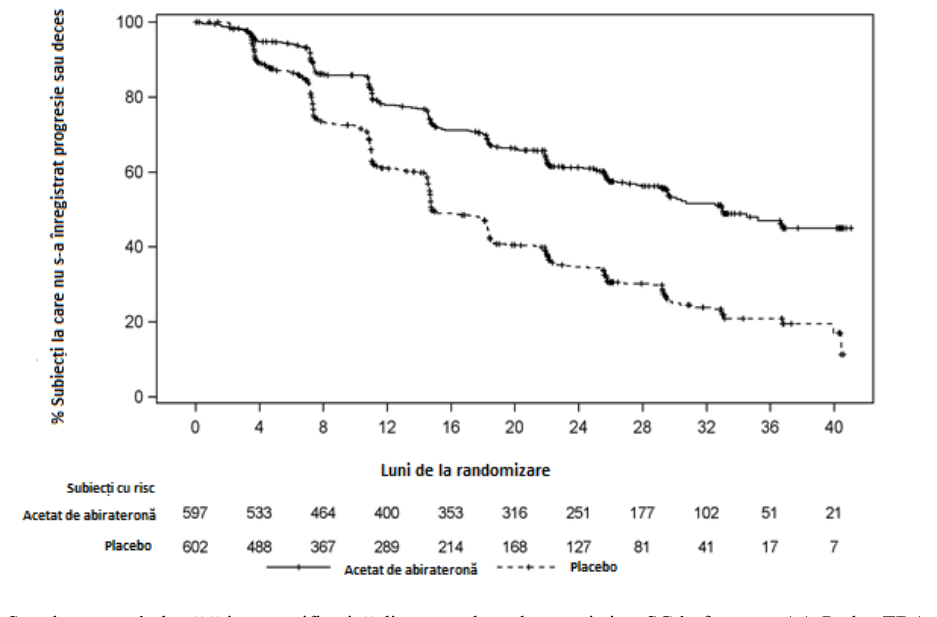
S-a observat o diferență semnificativă privind SFPr între grupurile de tratament (vezi Tabelul 2 și Figura 1).
Tabelul 2: Supraviețuire fără progresie radiologică - analiză stratificată; Populație cu intenție de tratament (Studiul PCR3011)
AA-P Placebo
Subiecți randomizați 597 602
Eveniment 239 (40,0%) 354 (58,8%)
Cenzurat 358 (60,0%) 248 (41,2%)
Timp până la eveniment
(luni)
Medie (IÎ 95%) 33,02 (29,57, NE) 14,78 (14,69, 18,27) Interval (0,0+, 41,0+) (0,0+, 40,6+)
Valoarea p a < 0,0001
Risc relativ (IÎ 95%)b 0,466 (0,394, 0,550)
Notă: += observație cenzurată, ne=neevaluabil. Progresia radiologică și decesul sunt luate în considerare în definirea evenimentului SFPR. AA-P= subiecții care au primit acetat de abirateronă și prednison. a valoarea p este obținută dintr-un test log-rank stratificat în funcție de scorul ECOG PS (0/1 sau 2) și leziunea viscerală (absentă sau prezentă). b Riscul relativ este obținut dintr-un model de riscuri proporționale stratificate. Risc relativ <1 este în favoarea AA-P.
Figura 1: Grafic Kaplan-Meier privind supraviețuirea fără progresie radiologică; populație cu intenție de tratare (Studiul PCR3011)
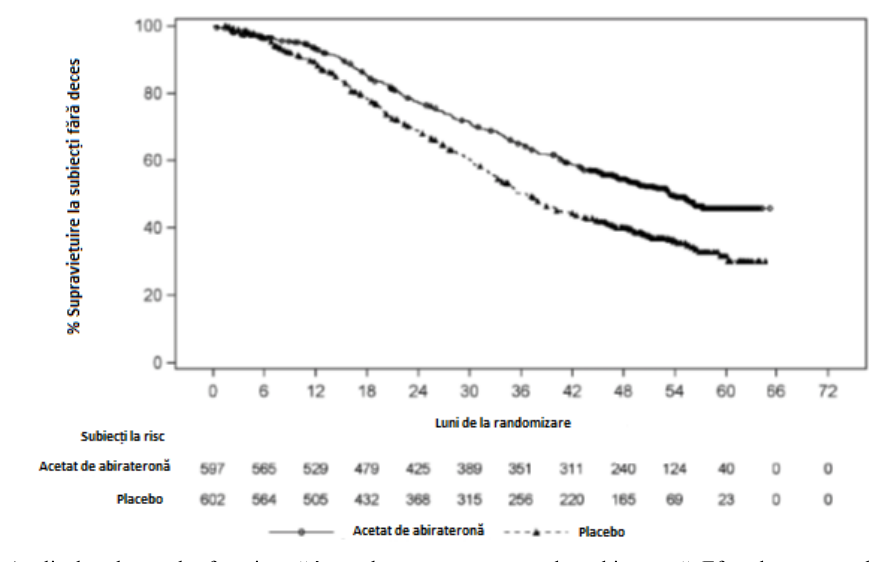
S-a observat o îmbunătățire semnificativă din punct de vedere statistic a SG în favoarea AA-P plus TDA, cu o reducere de 34% a riscului de deces în comparație cu placebo plus TDA (RR=0,66; IÎ 95%: 0,56, 0,78; p<0,0001) (vezi Tabelul 3 și Figura 2).
Tabelul Supravietuirea generală a pacientilor tratati fie cu abirateronă, fie cu
3 placebo în studiul PCR3011 (analiza intenției de tratare)
| Supraviețuirea globală | Abirateronă cu | Placebo | |
| Prednison (N=597) | (N=602) | ||
| Decese (%) | 275 (46%) | 343 (57%) | |
| Supraviețuire medie (luni) | 53,3 | 36.5 | |
| (IÎ 95%) | (48,2, NE) | (33.5, 40.0) | |
| Risc relativ ( IÎ 95%)1 | 0,66 (‘056’, ‘078’) | ||
| NE = Neevaluabil | |||
|---|---|---|---|
| 1 Riscul relativ este obținut dintr-un model de riscuri proporționale stratificate. Risc relativ <1 este în | |||
| favoarea abirateronei în asociere cu prednison. | |||
Figura 2: Grafic Kaplan-Meier privind supraviețuirea generală; analiza populației cu intenție de tratament în studiul PCR3011
Analizele subgrupelor favorizează în mod constant tratamentul cu abirateronă. Efectul tratamentului cu AA-P asupra SFPR și SG în cadrul subgrupurilor predeterminate a fost favorabil și a corespuns populației generale a studiului, cu excepția subgrupului cu scor ECOG de 2, unde nu s-a observat nicio tendință de beneficiu, însă dimensiunea redusă a eșantionului (n=40) limitează orice concluzie semnificativă.
Pe lângă îmbunătățirile observate în supraviețuirea globală și în SFPR, s-a demonstrat beneficiul asociat cu abiraterona față de tratamentul cu placebo în toate obiectivele finale secundare definite prospectiv.
Studiul STAMPEDE (pacienți cu mHSPC recent diagnosticat)
În studiul STAMPEDE, 901 pacienți cu mHSPC M1 au fost randomizați pentru a li se administra TPA ca monoterapie sau asocierea acetat de abirateronă (1000 mg zilnic) și prednisolon/prednison (5 mg zilnic; AAP) și au fost supuși stratificării utilizând criteriile de risc LATITUDE (Hoyle și colab. Eur Urol. 2019).
În total, 428 (48%) pacienți au fost clasificați ca având un risc scăzut conform criteriilor LATITUDE, iar afecțiunea cu nivel crescut de risc, folosind criteriile LATITUDE, a fost observată la 473 (52%) pacienți. Aproape toți pacienții (95%) au avut o afecțiune recent diagnosticată. Monitorizarea mediană a fost de 42 de luni. Asocierea dintre TPA și AAP a demonstrat un avantaj al supraviețuirii globale față de TPA ca monoterapie, și anume, atât în subgrupul cu risc scăzut, cât și în cel cu risc ridicat, per total, conform criteriilor LATITUDE (tabelul 4).
Tabelul 4: Supraviețuirea globală
| Stampede abirateronă Criteriile LATITUDE Toți pacienții M1 | TPA ca monoterapie Nr. de evenimente/ Nr. de pacienți | TPA+AAP Nr. de evenimente/ Nr. de pacienți | RR; IÎ 95% |
|---|---|---|---|
| Global | 195/452 | 135/449 | 0,61 (0,49-0,79) |
| Risc scăzut | 53/220 | 41/208 | 0,66 (0,44-0,98) |
| Risc crescut | 142/232 | 94/241 | 0,54 (0,41-0,70) |
Într-o analiză actualizată cu o monitorizare de 73 de luni (James et al. Int J Cancer. 2022), a fost confirmată îmbunătățirea supraviețuirii globale a tuturor pacienților cu neoplasm de prostată în stadiu metastazic, indiferent dacă boala lor era cu risc ridicat sau scăzut (RR global = 0,60 [IÎ 95%: 0,50-0,71]; RR risc scăzut = 0,54 [IÎ 95%: 0,40-0,74]; și RR risc ridicat = 0,54 [IÎ 95%: 0,43-0,69]).
HSPC în stadiu non-metastazic, cu nivel crescut de risc
Studiul STAMPEDE (pacienți cu HSPC în stadiu non-metastazic, cu nivel crescut de risc, recent diagnosticat)
În studiul STAMPEDE (Attard et al. Lancet. 2022), 914 pacienți cu cancer de prostată în stadiu nonmetastazic, cu nivel crescut de risc, au fost randomizați pentru a li se administra TPA ca monoterapie sau asocierea acetat de abirateronă (1000 mg zilnic) și prednisolon/prednison (5 mg zilnic; AAP). Pacienții au avut fie afecțiune cu ganglioni pozitivi (N1), fie afecțiune cu ganglioni negativi (N0), cu risc ridicat, local avansată (cu cel puțin două dintre următoarele: un stadiu tumoral T3 sau T4, un scor Gleason ≥8 și un nivel PSA ≥40 ng/mL). Radioterapia locală (conform ghidurilor locale) a fost obligatorie pentru boala N0 și încurajată pentru boala N1. TPA a fost administrată timp de trei ani și AAP timp de doi ani sau până la progresie, oricare dintre acestea a survenit mai întâi. Aproape toți pacienții (96%) au avut afecțiune recent diagnosticată. Dintre acești pacienți, în grupul de terapie combinată 55% au avut boală N0 și 39% boală N1, comparativ cu 56% și, respectiv, 41% în grupul de control. În grupul de terapie combinată, 82% dintre pacienți au fost planificați să primească radioterapie locală, comparativ cu 81% în grupul de control. Monitorizarea mediană a fost de 72 de luni. Asocierea de TPA și AAP (plus radioterapie) a demonstrat un avantaj în ceea ce privește supraviețuirea fără metastaze și supraviețuirea globală față de TPA ca monoterapie (plus radioterapie) (tabelul 5).
Tabelul 5: Supraviețuirea fără metastaze și supraviețuirea globală
| Tratament standard Nr. de evenimente/ Nr. de pacienți | Asocierea dintre ADT + AAP Nr. de evenimente/ Nr. de pacienți | RR; IÎ 95% | |
| Supraviețuirea fără metastaze | 183/455 | 111/459 | 0,54 (0,43-0,68) |
| Supraviețuirea globală | 142/455 | 95/459 | 0,63 (0,48-0,82) |
Neoplasm de prostată metastatic, rezistent la orhiectomie
Studiul 302 (pacienţi care nu au fost trataţi anterior cu chimioterapie)
Acest studiu a inclus pacienţi care nu au fost trataţi anterior cu chimioterapie şi care au fost asimptomatici sau uşor simptomatici şi la care chimioterapia nu a fost indicată din punct de vedere clinic. Un scor de 0-1 în Formularul de evaluare a durerii/Brief Pain Inventory-Short Form (BPI-SF) în ultimele 24 de ore a fost considerat asimptomatic, iar un scor de 2-3 a fost considerat uşor simptomatic.
În studiul 302, (n=1088) media de vârstă a pacienţilor incluşi a fost de 71 de ani pentru pacienţii trataţi cu abirateronă plus prednison sau prednisolon şi de 70 de ani pentru pacienţii la care s-a administrat placebo plus prednison sau prednisolon. Numărul de pacienţi trataţi cu abirateronă în funcţie de grupul rasial a fost: caucazieni 520 (95,4%), afro-americani 15 (2,8%), asiatici 4 (0,7%) şi alţii 6 (1,1%). Statusul de performanţă al Grupului Estic de Cooperare în Oncologie (ECOG) a fost 0 pentru 76% dintre pacienţi şi 1 pentru 24% dintre pacienţii din ambele braţe de tratament. Cincizeci la sută dintre pacienţi prezentau numai metastaze osoase, un procent suplimentar de 31% dintre pacienţi prezentau metastaze osoase şi ale ţesuturilor moi sau ale ganglionilor limfatici, iar 19% dintre pacienţi aveau doar metastaze ale ţesuturilor moi sau ale ganglionilor limfatici. Pacienţii cu metastaze viscerale au fost excluşi. Obiectivele finale coprincipale cu privire la eficacitate au fost supravieţuirea globală şi supravieţuirea fără progresie radiologică (SFPr). În plus faţă de evaluarea obiectivelor finale co-principale, beneficiul a fost, de asemenea, evaluat din punct de vedere al intervalului de timp până la utilizarea opioidelor pentru durerea din cancer, al intervalului de timp până la iniţierea chimioterapiei citotoxice, al intervalului de timp până la deteriorarea scorului de performanţă ECOG cu ≥ 1 punct şi al intervalului de timp până la progresia
PSA pe baza criteriilor Prostate Cancer Working Group-2 (PCWG2). Tratamentele din studiu au fost întrerupte la momentul progresiei clinice confirmate. De asemenea, tratamentele puteau fi întrerupte la momentul progresiei radiologice confirmate, la discreţia investigatorului.
Supravieţuirea fără progresie radiologică (SFPr) a fost evaluată utilizând studii de imagistică secvenţială, după cum sunt definite de criteriile PCWG2 (pentru leziuni osoase) şi criteriile modificate RECIST (Response Evaluation Criteria In Solid Tumors) (pentru leziuni ale ţesuturilor moi). Analiza SFPr a utilizat evaluarea radiologică a progresiei revizuită central.
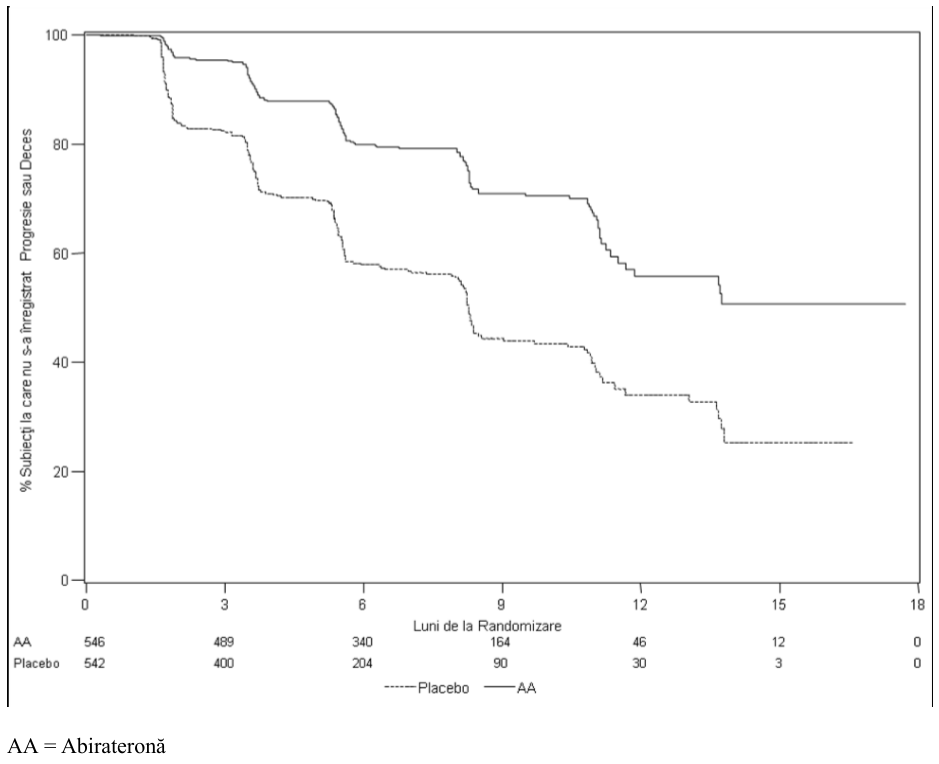
La analiza planificată a SFPr au existat 401 evenimente, 150 (28%) dintre pacienţii trataţi cu abirateronă şi 251 (46%) dintre pacienţii la care s-a administrat placebo prezentau dovezi radiologice ale progresiei sau decedaseră. S-a observat o diferenţă semnificativă între grupurile de tratament în ceea ce priveşte SFPr (vezi Tabelul 6 şi Figura 3).
Tabelul 6 Studiul 302: Supravieţuirea în absenţa progresiei radiologice la pacienţii la care s-a administrat fie abirateronă fie placebo în asociere cu prednison sau prednisolon plus analogi ai LHRH sau cu orhiectomie anterioară
| ABIRATERONĂ (N = 546) | Placebo (N = 542) | ||
| Supravieţuirea în absenţa | |||
|---|---|---|---|
| progresiei radiologice (SFPr) | |||
| Progresie sau deces | 150 (28%) | 251 (46%) | |
| Media SFPr exprimată în luni | Nu s-a atins | 8,3 | |
| (IÎ 95%) | (11,66; NE) | (8,12; 8,54) | |
| Valoare p * | < 0,0001 | ||
| Risc relativ** (IÎ 95%)b | 0,425 (0,347; 0,522) | ||
| NE= Neevaluabil. | |||
| * Valoarea p este obţinută dintr-un test log-rank stratificat în funcţie de scorul ECOG la momentul iniţial (0 sau 1) | |||
| **Risc relativ <1 este în favoarea abirateronei. | |||
Figura Curbele Kaplan Meier privind supravieţuirea în absenţa progresiei
3: radiologice la
pacienţii la care s-a administrat fie abirateronă, fie placebo, în asociere cu prednison sau prednisolon plus tratament cu analogi ai LHRH sau cu orhiectomie anterioară
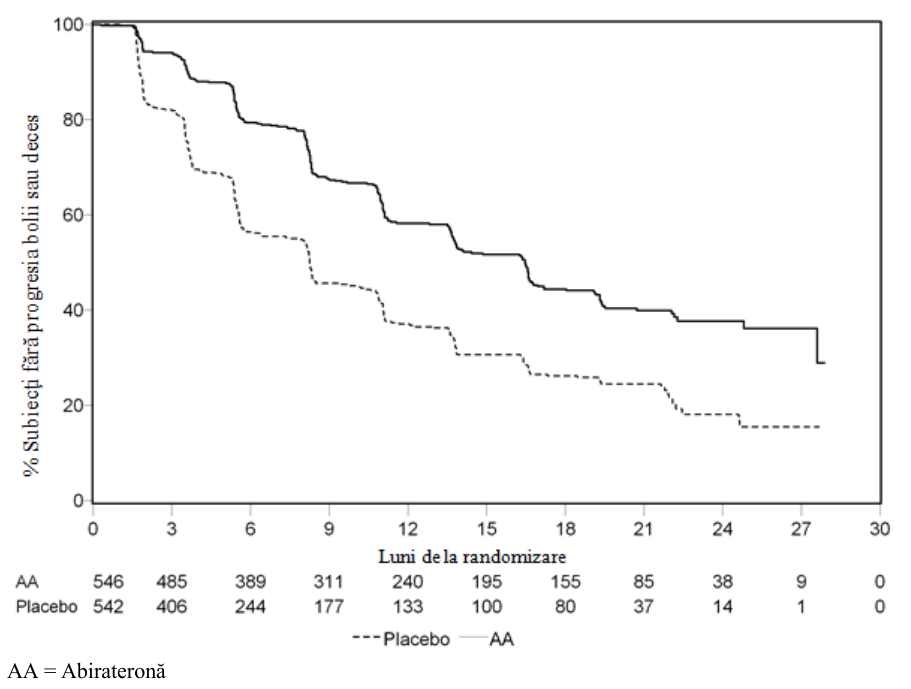
Cu toate acestea, datele subiecţilor s-au colectat în continuare până la data celei de-a doua analize interimare privind supravieţuirea globală (SG). Analiza radiologică a SFPr efectuată de investigator ca analiză de urmărire a sensibilităţii este prezentată în Tabelul 7 şi Figura 4.
Şase sute şapte (607) subiecţi prezentau progresie radiologică sau decedaseră: 271 (50%) din grupul de tratament cu acetat de abirateronă şi 336 (62%) din grupul placebo. Tratamentul cu acetat de abirateronă a redus riscul de progresie radiologică sau deces cu 47% în comparaţie cu placebo (HR=0,530; IÎ 95%: [0,451; 0,623], p<0,0001). Media rPFS a fost de 16,5 luni în grupul de tratament cu acetat de abirateronă şi de 8,3 luni la grupul placebo.
Tabelul Studiul 302: Supravieţuirea în absenţa progresiei radiologice la pacienţii la 7 care s-a
administrat fie abirateronă, fie placebo în asociere cu prednison sau prednisolon plus analogi ai LHRH sau cu orhiectomie anterioară (la a doua analiză interimară a SG- Analiza Investigatorului)
| ABIRATERONĂ (N = 546) | Placebo (N = 542) | ||
| Supravieţuirea în absenţa | |||
|---|---|---|---|
| progresiei radiologice (SFPr) | |||
| Progresie sau deces | 271 (50%) | 336 (62%) | |
| Media SFPr exprimată în luni | 16,5 | 8,3 |
|---|---|---|
| (IÎ 95%) | (13,80; 16,79) | (8,05; 9,43) |
| Valoarea p* | < 0,0001 | |
| Risc relativ** (IÎ 95%)b | 0,530 (0,451; 0,623) | |
| Media SFPr exprimată în luni | � 16,5 | 8,3 |
|---|---|---|
| (IÎ 95%) (13,80; 16,79) | (8,05; 9,43) | |
| Valoarea p* | � < 0,0001 | |
| Risc relativ** (IÎ 95%)b | � 0,530 (0,451; 0,623) | |
| Valoarea p este obţinută dintr-un test log-rank stratificat în funcţie de scorul ECOG la momentul sau 1) | ||
| **Risc relativ <1 este în favoarea abirateronei. | ||
Figura Curba Kaplan Meier privind supraviețuirea fără progresie radiologică la 4: pacienții tratați fie cu abirateronă, fie cu placebo în combinație cu
prednison sau prednisolon plus analogi ai LHRH sau orhiectomie anterioară (la a doua analiză intermediară a SG – analiza investigatorului)
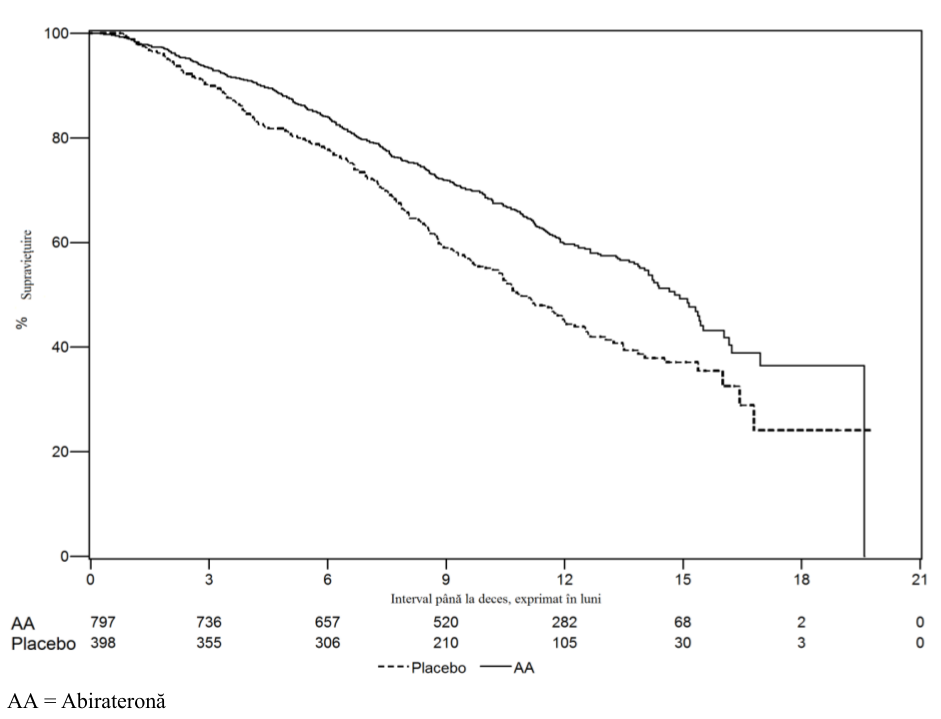
O analiză interimară (AI) planificată privind SG s-a efectuat după ce au fost observate 333 de decese. S-a renunţat la caracterul orb al studiului pe baza magnitudinii beneficiului clinic observat, iar pacienţilor din grupul placebo li s-a administrat tratament cu abirateronă. Supravieţuirea globală a fost mai mare pentru abirateronă decât pentru placebo, cu o reducere de 25% a riscului de deces (Rata de risc = 0,752; IÎ 95%: [0,606; 0,934], p=0.0097), dar SG nu a fost matură şi rezultatele interimare nu au întrunit valoarea pre-specificată de oprire pentru semnificaţie statistică (vezi Tabelul 4). A continuat să fie urmărită supravieţuirea şi după această AI.
Analiza finală planificată pentru SG s-a efectuat după ce au fost observate 741 decese (urmărirea medie a fost de 49 luni). Au decedat 65% (354 din 546) dintre pacienţii trataţi cu abirateronă comparativ cu 71% (387 din 542) dintre pacienţii trataţi cu placebo. A fost demonstrat un beneficiu semnificativ statistic al SG în favoarea grupului tratat cu abirateronă printr-o reducere cu 19,4% a riscului de deces (Rata de risc=0,806; IÎ 95%; [0,697; 0,931], p=0,0033) şi o îmbunătăţire a SG mediane de 4,4 luni (34,7 luni pentru abirateronă şi 30,3 luni pentru placebo) (vezi Tabelul 6 şi Figura 5). Această îmbunătăţire a fost demonstrată chiar dacă la 44% dintre pacienţii din braţul placebo au primit abirateronă ca tratament ulterior.
Tabelul Studiul 302: Supravieţuirea globală la pacienţii la care s-a administrat fie 8 abirateronă, fie placebo în asociere cu prednison sau prednisolon plus
analogi ai LHRH sau cu orhiectomie anterioară
| ABIRATERONĂ (N = 546) | Placebo (N = 542) | |||
| Analiza interimară a | ||||
|---|---|---|---|---|
| supravieţuirii | ||||
| Decese (%) | 147 (27%) | 186 (34%) | ||
| Media supravieţuirii (luni) | Nu s-a atins | 27,2 | ||
| (IÎ 95%) | (NE; NE) | (25,95; NE) | ||
| valoare p* | 0,0097 | |||
| Risc relativ** (IÎ 95%) | 0,752 (0,606; 0,934) | |||
| Analiza finală a supravieţuirii | ||||
| Decese | 354 (65%) | 387 (71%) | ||
| Media supravieţuirii în luni | 34,7 (32,7; 36,8) | 30,3 (28,7; 33,3) | ||
| (IÎ 95%) | ||||
| valoare p* | 0,0033 | |||
| Risc relativ** (IÎ 95%) | 0,806 (0,697; 0,931) | |||
| NE = Neevaluabil | ||||
| * Valoarea p este obţinută dintr-un test log-rank stratificat în funcţie de scorul ECOG la momentul iniţial (0 sau 1) | ||||
| **Risc relativ <1 în favoarea abirateronei. | ||||
Figura Curbele Kaplan Meier privind supravieţuirea la pacienţii la care s-a
5: administrat fie
abirateronă, fie placebo, în asociere cu prednison sau prednisolon plus tratament cu analogi ai LHRH sau cu orhiectomie anterioară, analiza finală
În plus faţă de îmbunătăţirile observate în ceea ce priveşte supravieţuirea globală şi SFPr, beneficiul abirateronei a fost demonstrat în comparaţie cu tratamentul cu placebo pentru toate măsurile obiectivelor finale secundare, după cum urmează:
Durata de timp până la progresia PSA pe baza criteriilor PCWG2: Media intervalului de timp până la progresia PSA a fost de 11,1 luni pentru pacienţii la care a fost administrat abirateronă şi de 5,6 luni pentru pacienţii la care a fost administrat placebo (RR=0,488; IÎ 95%: [0,420; 0,568], p < 0,0001). Durata de timp până la progresia PSA a fost aproximativ dublă în cazul tratamentului cu abirateronă (RR=0,488). Proporţia subiecţilor cu un răspuns confirmat PSA a fost mai mare în grupul abirateronă în comparaţie cu grupul placebo (62% comparativ cu 24%; p < 0,0001). La subiecţii cu boală măsurabilă a ţesuturilor moi, în cazul tratamentului cu abirateronă, au fost observate valori semnificativ crescute ale răspunsurilor tumorale complete şi parţiale.
Intervalul de timp până la utilizarea opioidelor pentru durerea din cancer: Media intervalului de timp până utilizarea opioidelor pentru durerea asociată neoplasmului de prostată la momentul analizei finale a fost de 33,4 luni pentru pacienţii la care s-a administrat abirateronă şi a fost de 23,4 luni pentru pacienţii la care s-a administrat placebo (HR=0,721; IÎ 95%: [0,614; 0,846], p<0,0001).
Intervalul de timp până la iniţierea chimioterapiei citotoxice: Media intervalului de timp până la iniţierea chimioterapiei citotoxice a fost de 25,2 luni pentru pacienţii la care s-a administrat abirateronă şi de 16,8 luni pentru pacienţii la care s-a administrat placebo (RR=0,580; IÎ 95%: [0,487; 0,691], p < 0,0001).
Intervalul de timp până la deteriorarea scorului de performanţă ECOG cu ≥ 1 punct: Media intervalului de timp până la deteriorarea scorului de performanţă ECOG cu ≥ 1 punct a fost de 12,3 luni pentru pacienţii la care s-a administrat abirateronă şi de 10,9 pentru pacienţii la care s-a administrat placebo (HR=0,821; IÎ 95%: [0,714; 0,943], p=0,0053.
Următoarele obiective finale ale studiului au demonstrat un avantaj statistic semnificativ în favoarea tratamentului cu abirateronă:
Răspuns obiectiv: Răspunsul obiectiv a fost definit ca procentul de subiecţi cu boală măsurabilă care au obţinut un răspuns complet sau parţial în conformitate cu criteriile RECIST (dimensiunea nodulilor limfatici la momentul iniţial trebuia să fie ≥ 2 cm pentru a fi consideraţi o leziune ţintă). Procentul de subiecţi cu boală măsurabilă la momentul iniţial care au înregistrat un răspuns obiectiv a fost de 36% în grupul abirateronă şi de 16% în grupul placebo (p < 0,0001).
Durere: Tratamentul cu abirateronă a redus semnificativ riscul de progresie a mediei intensităţii durerii cu 18% în comparaţie cu placebo (p=0,0490). Media intervalului de timp până la progresie a fost de 26,7 luni în grupul abirateronă şi de 18,4 luni în grupul placebo.
Intervalul de timp până la scăderea scorului FACT-P (Scor total): Tratamentul cu abirateronă a redus riscul de scădere a scorului FACT-P (Scor total) cu 22% în comparţie cu placebo (p=0,0028). Media intervalului de timp până la scăderea scorului FACT-P (Scor total) a fost de 12,7 luni în grupul abirateronă şi de 8,3 luni în grupul placebo.
Studiul 301 (pacienţi care au primit anterior chimioterapie)
Studiul 301 a inclus pacienţi la care a fost administrat anterior docetaxel. Nu a fost necesară demonstrarea progresiei bolii sub docetaxel, deoarece toxicitatea apărută în urma acestei chimioterapii ar fi putut determina întreruperea tratamentului. Pacienţii au continuat tratamentele din studiu până la înregistrarea unei progresii a valorilor PSA (creştere confirmată de 25% faţă de valoarea iniţială a pacientului/limita inferioară), împreună cu progresia radiologică şi progresia simptomatică sau clinică definite prin protocol. Pacienţii trataţi anterior cu ketoconazol pentru neoplasm de prostată au fost excluşi din acest studiu. Obiectivul final principal de evaluare a eficacității a fost supravieţuirea generală.
Vârsta medie a pacienţilor incluşi în studiu a fost de 69 de ani (interval 39-95 ani). Numărul pacienţilor trataţi cu abirateronă pe distribuţia rasială a fost de 737 (93,2%) de pacienţi caucazieni, 28
(3,5%) de pacienţi de culoare, 11 (1,4%) pacienţi asiatici şi 14 (1,8%) pacienţi aparţinând altor rase. Unsprezece la sută dintre pacienţii incluşi în studiu au avut un scor de performanţă ECOG de 2; 70% au avut dovezi radiologice de progresie a bolii, cu sau fără progresie a PSA; la 70% s-a administrat anterior un singur regim de chimioterapie citotoxică, iar la 30% s-au administrat două astfel de regimuri chimioterapice. Metastazele hepatice au fost prezente la 11% dintre pacienţii trataţi cu abirateronă.
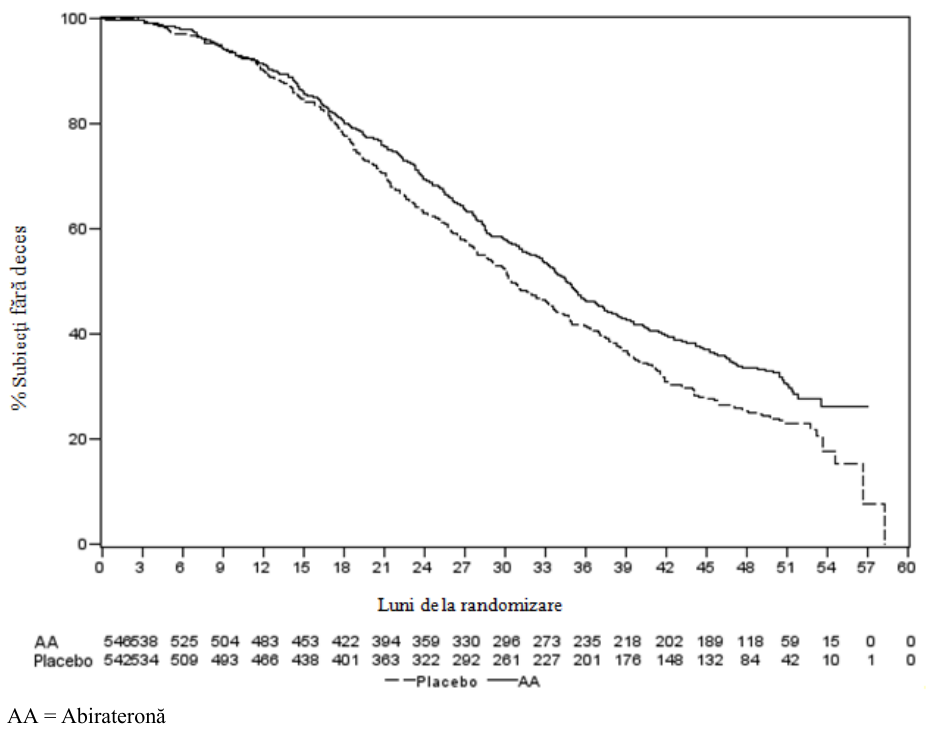
Într-o analiză planificată efectuată după ce au fost constatate 552 de decese, 42% (333 din 797) dintre pacienţii trataţi cu abirateronă decedaseră, comparativ cu 55% (219 din 398) dintre pacienţii la care s-a administrat placebo. La pacienţii trataţi cu abirateronă s-a observat o îmbunătăţire semnificativă statistic a supravieţuirii globale mediane (vezi Tabelul 7).
Tabelul Supravieţuirea generală la pacienţii la care s-a administrat fie
9 abirateronă, fie
placebo, în asociere cu prednison sau prednisolon plus tratament cu analogi ai
LHRH sau cu orhiectomie anterioară
| ABIRATERONĂ (N = 797) | Placebo (N = 398) | ||
| Analiza principală privind | |||
|---|---|---|---|
| supravieţuirea Decese (%) Supravieţuirea medie (luni ) (IÎ 95%) | 333 (42%) 14,8 (14,1; 15,4) | 219 (55%) 10,9 (10,2; 12,0) |
|---|---|---|
| valoare pa Risc relativ (IÎ 95%)b | < 0,0001 0,646 (0,543; 0,768) | |
| Analiza actualizată privind supravieţuirea Decese (%) Supravieţuirea medie (luni) (IÎ 95%) | 501 (63%) 15,8 (14,8; 17,0) | 274 (69%) 11,2 (10,4; 13,1) |
| Risc relativ (IÎ 95%)b | 0,740 (0,638; 0,859) | |
| aValoarea p provine dintr-un test logaritmic stratificat în funcţie de scorul statusului de performanţă conform ECOG (0- | ||
1 faţă de 2), scorul pentru durere (absență faţă de prezență), numărul de tratamente chimioterapice anterioare (1 faţă de
2), şi tipul de progresie a bolii (numai PSA faţă de radiologică). b Riscul relativ provine dintr-un model de riscuri proporţionale stratificate. Riscul relativ < 1 este în favoarea abirateronei.
La toate punctele din timp la care s-a realizat evaluarea după primele câteva luni de la iniţierea tratamentului, un procent mai mare de pacienţi trataţi cu abirateronă au rămas în viaţă, comparativ cu procentul de pacienţi la care s-a administrat placebo (vezi Figura 6).
Figura Curbele Kaplan Meier privind supravieţuirea pacienţilor la care s-a
6: administrat fie abirateronă, fie placebo, în asociere cu prednison sau
prednisolon plus tratament cu analogi ai LHRH sau cu orhiectomie anterioară
Analizele privind supravieţuirea pe subgrupe au demonstrat un beneficiu semnificativ privind supravieţuirea pentru tratamentul cu abirateronă (vezi Figura 7).
Figura 7: Supravieţuirea generală pe subgrupe: risc relativ şi interval de încredere 95%
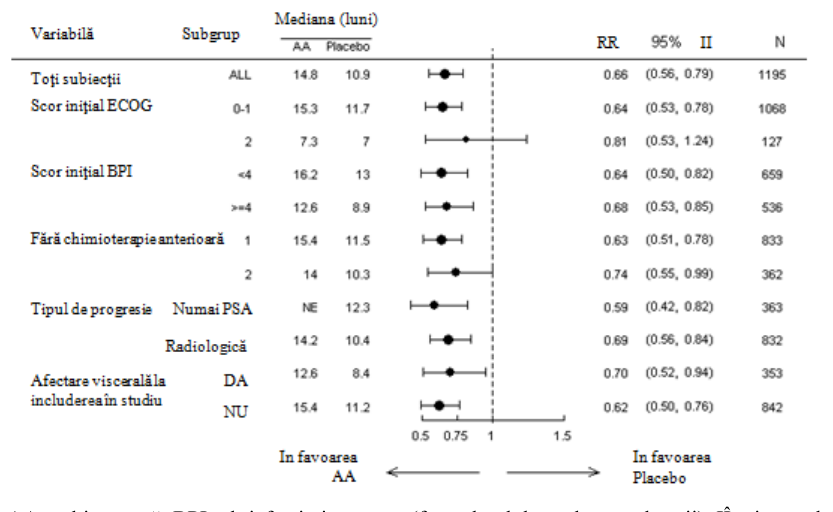
AA = abirateronă; BPI = brief pain inventory (formularul de evaluare a durerii); IÎ = interval de încredere; ECOG = scor de performanţă conform Eastern Cooperative Oncology Group; RR=risc relativ; NE = neevaluabil
În plus faţă de îmbunătăţirea observată în ceea ce priveşte supravieţuirea globală, toate criteriile de evaluare finale secundare ale studiului au favorizat abiraterona şi au fost semnificative statistic după ajustarea pentru teste multiple, după cum urmează:
Pacienţii cărora li s-a administrat abirateronă au demonstrat o rată totală de răspuns a PSA (definită ca o reducere de ≥50% faţă de valoarea iniţială) semnificativ mai mare comparativ cu pacienţii la care s-a administrat placebo, 38% faţă de 10%, p < 0,0001.
Durata medie de progresie a PSA a fost de 10,2 luni pentru pacienţii trataţi cu abirateronă şi de 6,6 luni pentru pacienţii la care s-a administrat placebo (RR = 0,580; IÎ 95%: [0,462;0,728], p < 0,0001).
Supravietuirea medie fără progresie radiologică a fost de 5,6 luni pentru pacienţii trataţi cu abirateronă şi de 3,6 luni pentru pacienţii la care s-a administrat placebo (RR = 0,673; IÎ 95%: [0,585; 0,776], p < 0,0001).
Durerea
Procentul de pacienţi care au resimţit ameliorarea durerii a fost semnificativ statistic mai mare în grupul tratat cu abirateronă comparativ cu grupul placebo (44% faţă de 27%, p = 0,0002). Un răspuns la tratament a fost definit ca un pacient care a înregistrat în ultimele 24 ore o reducere de minimum 30% faţă de valoarea iniţială a celui mai mare scor BPI-SF de evaluare a intensităţii durerii, fără nicio creştere a scorului de utilizare a analgezicelor, observată la două evaluări consecutive efectuate la interval de patru săptămâni. Numai pacienţii cu un scor de durere iniţial ≥ 4 şi cei cu cel puţin încă un scor de evaluare a durerii după valoarea iniţială (N = 512) au fost evaluaţi în ceea ce priveşte ameliorarea durerii.
La 6 luni (22% faţă de 28%), 12 luni (30% faţă de 38%) şi 18 luni (35% faţă de 46%), un procent mai mic de pacienţi trataţi cu abirateronă a înregistrat o progresie a durerii comparativ cu pacienţii la care s-a administrat placebo. Progresia durerii a fost definită ca o creştere în ultimele 24 ore a celui mai mare scor BPI-SF de evaluare a intensităţii durerii ≥ 30% faţă de valoarea iniţială, fără nicio scădere a scorului de utilizare a analgezicelor, observată la două evaluări consecutive, sau o creştere ≥ 30% a scorului de utilizare a analgezicelor observată la două vizite consecutive. Timpul până la progresia durerii la percentila 25 a fost de 7,4 luni în grupul tratat cu abirateronă, comparativ cu 4,7 luni în grupul placebo.
Evenimente osoase asociate
La un procent mai mic de pacienţi din grupul tratat cu abirateronă s-au înregistrat evenimente osoase, comparativ cu grupul placebo la 6 luni (18% faţă de 28%), 12 luni (30% faţă de 40%) şi la 18 luni (35% faţă de 40%). Timpul până la primul eveniment musculo-scheletic la percentila 25 din grupul tratat cu abirateronă fost dublu faţă de grupul de control, şi anume 9,9 luni comparativ cu 4,9 luni. Un eveniment osos a fost definit ca fiind o fractură patologică, compresie medulară, radioterapie paliativă la nivelul osului, sau intervenţii chirurgicale la nivel osos.
Copii şi adolescenţi
Agenţia Europeană a Medicamentului a acordat o derogare de la obligaţia de depunere a rezultatelor studiilor efectuate cu produsul de referință care conține abirateronă la toate subgrupele de copii şi adolescenţi cu neoplasm de prostată în stadiu avansat. Vezi pct. 4.2 pentru informaţii privind utilizarea la copii şi adolescenţi
5.2 Proprietăţi farmacocinetice
După administrarea de acetat de abirateronă, farmacocinetica abirateronei şi acetatului de abirateronă a fost studiată la subiecţi sănătoşi, la pacienţi cu neoplasm de prostată metastatic în stadiu avansat şi la subiecţi fără neoplazii, dar cu insuficienţă hepatică sau renală. Acetatul de abirateronă este rapid convertit in vivo în abirateronă, un inhibitor al biosintezei de hormoni androgeni (vezi pct. 5.1).
Absorbţie
După administrarea orală de acetat de abirateronă în condiţii de repaus alimentar, timpul până la atingerea concentraţiei plasmatice maxime de abirateronă este de aproximativ 2 ore.
Administrarea de acetat de abirateronă împreună cu alimente, în comparaţie cu administrarea în condiţii de repaus alimentar, are ca rezultat o creştere de până la de 10 ori (ASC) şi de până la de 17 ori (Cmax) a expunerii sistemice medii la abirateronă, în funcţie de conţinutul de grăsimi al alimentelor. Având în vedere variaţia normală a conţinutului şi compoziţiei alimentelor la o masă, administrarea de abirateronă în timpul mesei poate determina expuneri foarte variabile. Prin urmare, abiraterona nu trebuie administrată împreună cu alimente. Trebuie administrat cu cel puţin o oră înainte de masă sau la cel puțin două ore după masă. Comprimatele trebuie înghiţite întregi, cu apă (vezi pct. 4.2).
Distribuţie
La om, legarea abirateronei marcată cu 14C de proteinele plasmatice este de 99,8%. Volumul aparent de distribuţie este de aproximativ 5630 l, sugerând că abiraterona se distribuie în proporţie mare în ţesuturile periferice.
Metabolizare
După administrarea orală de acetat de abirateronă marcată cu 14C, sub formă de capsule, acetatul de abirateronă este hidrolizat la abirateronă, care este supusă ulterior metabolizării, incluzând sulfatare, hidroxilare şi oxidare, în principal, la nivel hepatic. Majoritatea radioactivităţii circulante (aproximativ 92%) se găseşte sub formă de metaboliţi ai abirateronei. Din 15 metaboliţi detectabili, doi metaboliţi principali, sulfatul de abirateronă şi N-oxidul sulfatului de abirateronă, fiecare reprezintă aproximativ 43% din radioactivitatea totală.
Eliminare
Timpul mediu de înjumătăţire plasmatică prin eliminare a abirateronei este de aproximativ 15 ore pe baza datelor provenite de la subiecţi sănătoşi. După administrarea orală a 1000 mg acetat de abirateronă marcată cu 14C, aproximativ 88% din doza radioactivă se regăseşte în materiile fecale şi aproximativ 5% în urină. Principalii compuşi prezenţi în materiile fecale sunt acetatul de abirateronă şi abiraterona nemodificată (aproximativ 55% şi, respectiv 22% din doza administrată).
Insuficienţă hepatică
Farmacocinetica acetatului de abirateronă a fost studiată la subiecţi cu insuficienţă hepatică, uşoară sau moderată (Clasa A şi respectiv B conform clasificării Child-Pugh) preexistentă şi la subiecţii sănătoşi incluşi în grupul de control. Expunerea sistemică la abirateronă după administrarea unei doze orale unice de 1000 mg a crescut cu aproximativ 11% şi 260% la subiecţii cu insuficienţă hepatică preexistentă uşoară, respectiv moderată. Timpul mediu de înjumătăţire plasmatică prin eliminare a abirateronei este prelungit la aproximativ 18 ore la subiecţii cu insuficienţă hepatică uşoară şi la aproximativ 19 ore la subiecţii cu insuficienţă hepatică moderată.
În cadrul unui alt studiu clinic a fost evaluat profilul farmacocinetic al abirateronei la subiecţii cu insuficienţă hepatică severă preexistentă (n=8) (Clasa C conform clasificării Child-Pugh) şi la 8 subiecţi sănătoşi, martori cu funcţie hepatică normală. ASC a abirateronei a crescut cu aproximativ 600%, iar fracţia liberă a medicamentului a crescut cu 80% la subiecţii cu insuficienţă hepatică severă comparativ cu subiecţii care au prezentat funcţie hepatică normală.
Nu este necesară ajustarea dozei la pacienţii cu insuficienţă hepatică uşoară preexistentă. Utilizarea acetatului de abirateronă trebuie evaluată cu atenţie la pacienţii cu insuficienţă hepatică moderată astfel încât beneficiile să depăşească posibilele riscuri (vezi pct. 4.2 şi 4.4). Acetatul de abirateronă nu trebuie utilizat la pacienţii cu insuficienţă hepatică severă (vezi pct. 4.2, 4.3 şi 4.4).
La pacienţii care dezvoltă hepatotoxicitate în timpul tratamentului, poate fi necesară întreruperea tratamentului şi ajustarea dozei (vezi pct. 4.2 şi 4.4).
Insuficienţă renală
Farmacocinetica acetatului de abirateronei a fost comparată la pacienţii cu boală renală în stadiu terminal, incluşi într-un program stabil de hemodializă cu farmacocinetica la subiecţii cu funcţie renală normală, incluşi în grupul de control. După administrarea unei doze orale unice de 1000 mg, expunerea sistemică la abirateronă nu a crescut la subiecţii cu boală renală în stadiu terminal care efectuează şedinţe de dializă. Administrarea la pacienţii cu insuficienţă renală, inclusiv insuficienţă renală severă, nu necesită reducerea dozei (vezi pct. 4.2). Cu toate acestea, nu există experienţă clinică la pacienţii cu neoplasm de prostată şi insuficienţă renală severă. Se recomandă prudenţă la aceşti pacienţi.
5.3 Date preclinice de siguranţă
În toate studiile privind evaluarea toxicităţii la animale, concentraţiile de testosteron circulant au fost reduse semnificativ. Ca urmare, s-a observat scăderea în greutate a organelor şi modificări morfologice şi/sau histopatologice la nivelul organelor de reproducere şi al glandelor suprarenale, hipofizei şi glandei mamare. Toate modificările au demonstrat reversibilitate completă sau parţială.
Modificările la nivelul organelor de reproducere şi organelor sensibile la hormoni androgeni sunt în concordanţă cu farmacologia abirateronei. Toate modificările hormonale asociate tratamentului au dispărut ori s-au rezolvat după o perioadă de recuperare de 4 săptămâni.
În cadrul studiilor pentru evaluarea fertilităţii atât la masculi, cât şi la femele de şobolan, acetatul de abirateronă a avut un efect de reducere a fertilităţii, care a fost complet reversibil după 4 - 16 săptămâni de la oprirea tratamentului cu acetat de abirateronă.
În cadrul unui studiu privind evaluarea toxicităţii asupra dezvoltării la şobolan, acetatul de abirateronă a afectat sarcina, inclusiv a redus greutatea fetală şi supravieţuirea. Cu toate că au fost observate efecte asupra organelor genitale externe, acetatul de abirateronă nu a fost teratogen.
În toate aceste studii privind toxicitatea asupra fertilităţii şi dezvoltării efectuate la şobolan, toate efectele au fost corelate cu activitatea farmacologică a abirateronei.
Cu excepţia modificărilor la nivelul organelor de reproducere observate în toate studiile privind evaluarea toxicităţii la animale, datele non-clinice nu au evidenţiat niciun risc special pentru om pe baza studiilor farmacologice convenţionale privind evaluarea siguranţei, toxicităţii după doze repetate, genotoxicităţii și a potențialului carcinogen. Acetatul de abirateronă nu a prezentat un potenţial carcinogenic într-un studiu cu durata de 6 luni la şoareci transgenici (Tg.rasH2). Într-un studiu privind carcinogenicitatea cu durata de 24 luni la şobolan, acetatul de abirateronă a crescut incidenţa neoplasmelor celulelor interstiţiale de la nivelul testiculelor. Se consideră că acest rezultat este asociat cu acţiunea farmacologică a abirateronei şi specifică şobolanului. Acetatul de abirateronă nu a fost carcinogenic la femelele şobolan.
Substanța activă, abiraterona, prezintă un risc pentru mediul acvatic, în special pentru pești.
6. PROPRIETĂŢI FARMACEUTICE
6.1 Lista excipienţilor
Nucleu
Croscarmeloză sodică
Laurilsulfat de sodiu
Povidonă (E1201)
Celuloză microcristalină (E460)
Lactoză monohidrat
Dioxid de siliciu coloidal anhidru (E551)
Stearat de magneziu (E470b)
Film
Alcool polivinilic (E1203)
Dioxid de titan (E171)
Macrogol (E1521)
Talc (E553b)
Oxid roșu de fer (E172)
Oxid negru de fer (E172)
6.2 Incompatibilităţi
Nu este cazul.
6.3 Perioada de valabilitate
Blistere: 3 ani
Flacoane: 3 ani
6.4 Precauţii speciale pentru păstrare
Acest medicament nu necesită condiții speciale de păstrare.
6.5 Natura şi conţinutul ambalajului
Comprimatele filmate sunt furnizate în:
- Blistere din aluminiu-OPA/Al/PVC sau aluminiu-PVC/PE/PVDC conținând 56, 60, 84, 112 comprimate filmate.
- Blistere din aluminiu-OPA/Al/PVC sau aluminiu-PVC/PE/PVDC perforate unidoză conținând 56 x 1, 60 x 1, 84 x 1, 112 x 1 comprimate filmate.
- Flacoane din polietilenă de înaltă densitate (PEÎD), cu dispozitiv de absorbție de oxigen, închise cu capac din polipropilenă (PP) prevăzut cu sistem de siguranță pentru copii, conținând 60 comprimate filmate.
Este posibil ca nu toate mărimile de ambalaj să fie comercializate.
6.6 Precauţii speciale pentru eliminarea reziduurilor
Orice medicament neutilizat sau material rezidual trebuie eliminat în conformitate cu reglementările locale. Acest medicament poate reprezenta un risc pentru mediul acvatic (vezi pct. 5.3).
7. DEŢINĂTORUL AUTORIZAŢIEI DE PUNERE PE PIAŢĂ
Sandoz Pharmaceuticals S.R.L.
Calea Floreasca, nr. 169A
Clădirea A, etaj 1, sector 1, 014459, București, România
8. NUMĂRUL(ELE) AUTORIZAŢIEI DE PUNERE PE PIAŢĂ
16461/2026/01-17
9. DATA PRIMEI AUTORIZĂRI SAU A REÎNNOIRII AUTORIZAŢIEI
Data primei autorizări: Iulie 2021
Data ultimei reînnoiri a autorizaţiei: Februarie 2026
10. DATA REVIZUIRII TEXTULUI
Februarie 2026